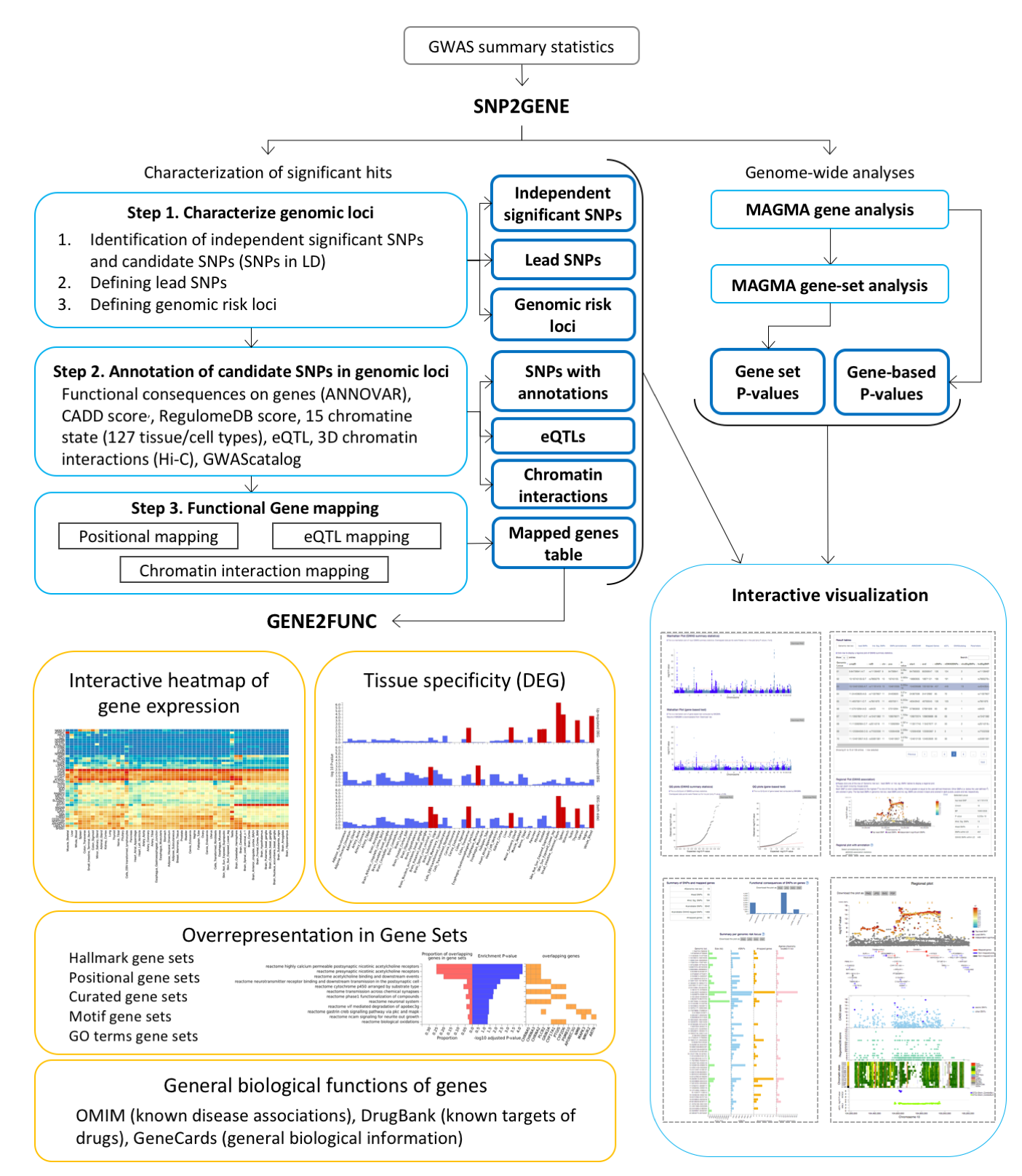
General overview of FUMAGWAS
The main purpose of FUMA is to use functional and biological information to prioritize genes based on GWAS outcomes.
FUMA consists of three modules: SNP2GENE, GENE2FUNC, and Cell Type.
To annotate and prioritize SNPs and genes from your GWAS summary statistics, go to SNP2GENE which compute LD structure, annotates functions to SNPs, and prioritize candidate genes.
You can then use the prioritized genes as input to GENE2FUNC to check expression patterns and shared molecular functions between genes. GENE2FUNC can also be used for any list of pre-selected genes (i.e. created outside of SNP2GENE).
To identify cell types that could be relevant for your GWAS summary statistics, you can run the Cell Type module.

Quick Start
General Information
Each page contains information where needed and brief descriptions of inputs and results to help you understand them without going through entire tutorial.
:click the question mark to display a brief description.
This is for optional inputs/parameters.
This is the message if everything is fine.
This is the message if the input/parameter is mandatory and not given or invalid input is given.
This is the warning message for the input/parameter. Please check your input settings.
Prioritize genes based on your own GWAS summary statistics
For risk loci identified by FUMA in your summary statistics, you can obtain functional annotation of SNPs and map them to genes. By changing parameter settings, you can control which annotations or filters need to be used to prioritize genes.
Because you will upload your own GWAS summary statistics, we require you to register. All uploaded files are handled securely and can only be seen by you. Results can be queried at later times, but can also be deleted. If you delete a previously run job, your uploaded file will be deleted from the FUMA server.
1. Registration/Login
If you haven't registered yet, please do so from Register.Before you submit your GWAS summary statistics, please log in to your account. You can login from either login page or SNP2GENE page directly.
2. Submit new job at SNP2GENE
A new job starts with a GWAS summary statistics file. A variety of file formats are supported. Please refer the section of Input files for details. If your input file is an output from PLINK, SNPTEST or METAL, you can directly submit the file without specifying column names.The input GWAS summary statistics file could be a subset of SNPs (e.g. only SNPs which are interesting in your study), but in this case, MAGMA results are not relevant anymore.
Optionally, if you would like to pre-specify lead SNPs, you can upload a file with 3 columns; rsID, chromosome and position. FUMA will then use these SNPs to select LD-related SNPs for annotation and mapping, instead of using lead SNPs identified by FUMA. If you want FUMA to analyze pre-specified lead SNPs, you will need to disable (uncheck) the section "Identify additional independent lead SNPs". Please note that in order for the pre-specified lead SNPs to be processsed by FUMA, they would need to be present in your input GWAS summary statistics file.
In addition, if you are interested in specific genomic regions, you can also provide them by uploading a file with 3 columns; chromosome, start, and end position. FUMA will then use these genomic regions to select LD-related SNPs for annotation and mapping, instead of determining the regions itself.
3. Set parameters
On the page where you upload the input files, there are a variety of optional parameters that control the prioritization of genes. Please check your parameters carefully. The default settings are to perform identification of independent genome-wide significant SNPs at r2 0.6 and lead SNPs at r2 0.1, and to maps SNPs to genes up to 10kb apart.To filter SNPs by specific functional annotations and to use eQTL mapping, please change parameters (please refer the parameter section of this tutorial from here).
If all inputs are valid, 'Submit Job' button will be activated. Once you submit a job, this will be listed in My Jobs.
Please do not navigate away from the page while your file is uploading (this may take up to couple of minutes depending on the file size and your internet speed).
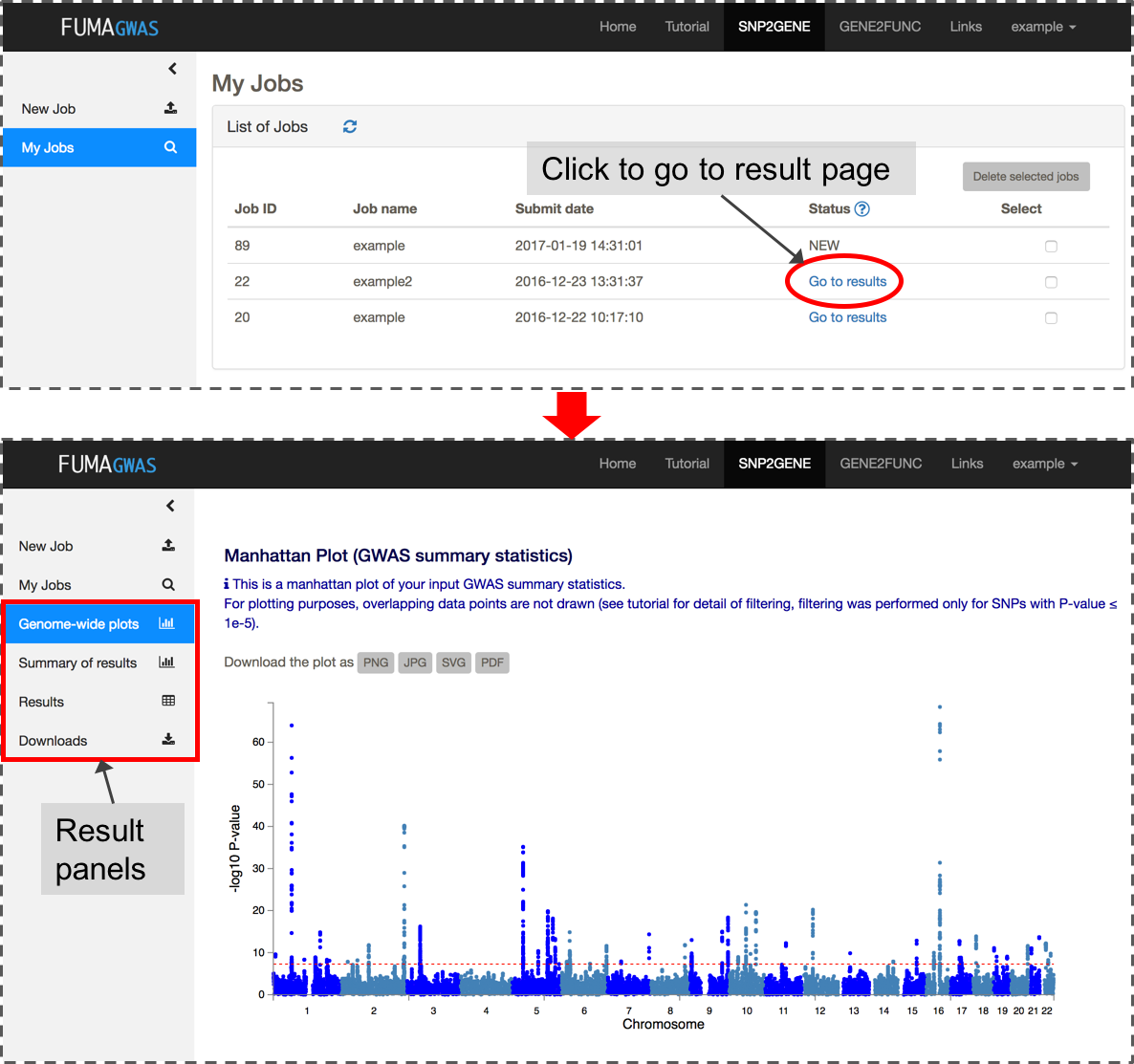
4. Check your results
After you submit files and parameter settings, a JOB has the status NEW which will be updated to QUEUES to RUNNING. Depending on the number of significant genomic regions, this may take between a couple of minutes and an hour. Once a JOB has finished running, you will receive an email. Unless an error occurred during the process, the email includes the link to the result page (this again requires login). You can also access the results page from My Jobs page.The result page displays 4 additional side bars.
Genome-wide plots: Manhattan plots and Q-Q plots for GWAS summary statistics and gene-based test by MAGMA, results of MAGMA gene-set analysis and tissue expression analysis.
Summary of results: Summary of results such as the number of lead and LD-related SNPs, and mapped genes for overall and per identified genomic risk locus.
Results: Tables of lead SNPs, genomic risk loci, candidate SNPs with annotations, eQTLs (only when eQTL mapping is performed), mapped genes and GWAS-catalog reported SNPs matched with candidate SNPs. You can also create interactive regional plots with functional annotations from this tab.
Downloads: Download all results as text files.
Details of all FUMA outputs are provided in the SNP2GENE Outputs section of this tutorial.
Gene functions: Tissue specific gene expression and shared biological functions of a list of genes
GENE2FUNC can take the list of prioritized genes from SNP2GENE or alternatively you can provide another list of pre-specified genes. Note that the genes prioritized in SNP2GENE are based on the functional and/or eQTL mapping, but not on MAGMA based gene output.
For every input genes, GENE2FUNC provides information on expression in different tissue types,
tissue specificity and enrichment of publicly available gene sets.
1. Submit a list of genes
Both a list of genes of interest and background genes (for hypergeometric test) are mandatory input.You can use mapped genes from SNP2GENE by clicking the "Submit" button in the result page (Results tab).
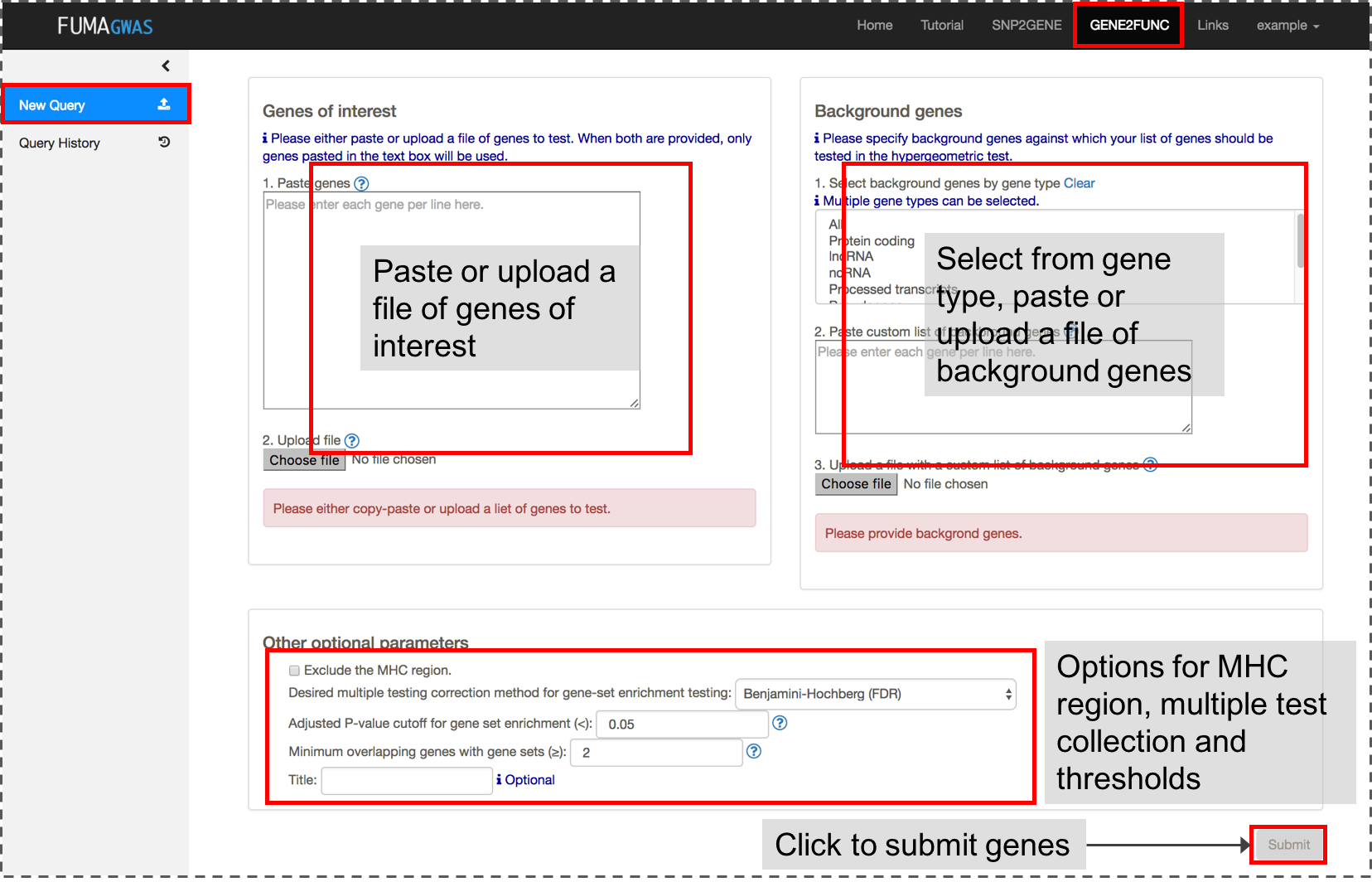
2. Results
Once genes are submitted, four extra side bars are shown.Gene Expression: An interactive heatmap of gene expression of user selected data sets.
Tissue Specificity: Bar plots for enrichment test of differentially expressed genes in a certain label compared to all other samples for a use selected data sets. See GENE2FUNC Outputs section for details.
Gene Sets: Plots and tables of enriched gene sets.
Gene Table: Table of input genes with links to external databases; OMIM, Drugbank and GeneCards.
Further details are provided in the GENE2FUNC Outputs section of this tutorial.
SNP2GENE
Prepare Input Files
1. GWAS summary statistics
GWAS summary statistics is a mandatory input of SNP2GENE process.
FUMA accepts various types of format. For example, PLINK, SNPTEST and METAL output formats can be used as is.
For other formats, column names can be provided.
Input files should be prepared in ascii txt or (preferably) gzipped or zipped.
Every row should contain information on one SNP.
An input GWAS summary statistics file could contain only subset of SNPs (e.g. SNPs of interest for your study to annotate them),
but in this case, results of MAGMA will not be relevant anymore.
Please note that variants that do not exist in the selected reference panel will not be included in any analyses. The 1000G reference panel is provided in the Download page (scroll to the section Reference panel data).
For indels, both alleles need to be matched exactly with reference panel to be included in the analysis.
For example, an indel rs144029872 needs to be encoded with AG/A (the order of alleles does not matter),
anything else such as G/- or I2/D will not match with the selected reference panel.
Input Build
The reference data included in FUMA SNP2GENE is on build GRCh37 (hg19).
If your data is build GRCh37, you can upload your file.
If your data is build GRCh38, you can upload your file if it includes rsIDs and does not include chromosome and position columns.
As of FUMA v1.6.0, there is now the option to upload data with build GRCh38 if it includes columns for chromosome, position, effect allele, and noneffect allele.
If your input data includes all of these columns and you select "Input is build GRCh38" in the submission page, then your data will be annotated with rsIDs based on dbSNP v150 data.
Only variants included in the user selected reference panel ("Reference panel population") will be given an rsID.
The variants that could not be given rsIDs can be downloaded after your job is completed (GRCh38_droppedvariants.txt.gz).
Mandatory columns
The input file must include a P-value and either an rsID or chromosome index + genetic position on hg19 reference genome.
When either chromosome or position is missing, they are extracted from dbSNP build 146 based on rsID.
In this case, input rsID is updated to dbSNP build 146.
When rsID is missing, it is extracted from dbSNP build 146 based on chromosome and position.
The column of chromosome can be a string such as "chr1" or just an integer such as 1.
When "chr" is attached, this will be removed in output files.
When the input file contains chromosome X, this will be encoded as chromosome 23, however, the input file can contain "X".
Allele columns
Alleles are not mandatory but if only one allele is provided, that is considered to be the effect allele.
When two alleles are provided, the effect allele will be defined depending on column name.
If alleles are not provided, they will be extracted from the dbSNP build 146 and minor alleles will be assumed to be the effect alleles.
Effect and non-effect alleles are not distinguished during annotations, but used for alignment with eQTLs.
Whenever alleles are provided, they are matched with dbSNP build 146 if extraction of rsID, chromosome or position is necessary.
Alleles are case insensitive.
Headers
Column names are automatically detected based on the following headers (case insensitive).
- SNP | snpid | markername | rsID: rsID
- CHR | chromosome | chrom: chromosome
- BP | pos | position: genomic position (hg19)
- A1 | effect_allele | allele1 | alleleB: affected allele
- A2 | non_effect_allele | allele2 | alleleA: another allele
- P | pvalue | p-value | p_value | pval: P-value (Mandatory)
- OR: Odds Ratio
- Beta | be: Beta
- SE: Standard error
Extra columns will be ignored.
Rows that start with "#" will be ignored.
Column "N" is described in the Parameters section.
Be careful with the alleles header in which A1 is defined as effect allele by default. Please specify both effect and non-effect allele column to avoid mislabeling.
If wrong labels are provided for alleles, it does not affect any annotation and prioritization results. It does however affect eQTLs results (alignment of risk increasing allele of GWAS and tested allele of eQTLs). Be aware of that when you interpret results.
Delimiter
Delimiter can be any of white space including single space, multiple space and tab.
Because of this, each element including column names must not include any space.
Note and Tips
When the input file has all of the following columns; rsID, chromosome, position, allele1 and allele2, the process will be much quicker than extracting information.
The pipeline currently supports human genome hg19. If your input file is not based on hg19, please update the genomic position using liftOver from UCSC. However, there is an option for you!! When you provide only rsID without chromosome and genomic position, FUMA will extract them from dbSNP build 146 based on hg19. To do this, remove columns of chromosome and genomic position or rename headers to ignore those columns. Note that extracting chromosome and genomic position will take extra time.
2. Pre-defined lead SNPs
This is an optional input file.
This option would be useful when
1. You have lead SNPs of interest but they do not reach significant P-value threshold.
2. You are only interested in specific lead SNPs and do not want to identify additional lead SNPs which are independent.
In this case, you also have to UNCHECK option of Identify additional independent lead SNPs.
If you want to specify lead SNPs, input file should have the following 3 columns:
- rsID : rsID of the lead SNPs
- chr : chromosome
- pos : genomic position (hg19)
The order of columns has to be exactly the same as shown above but header could be anything (the first row is ignored). Extra columns will be ignored.
3. Pre-defined genomic region
This is an optional input file.
This option would be useful when you have already done some follow-up analyses of your GWAS and are interested in specific genomic regions.
When pre-defined genomic region is provided, regardless of parameters, only lead SNPs and SNPs in LD with them within provided regions will be reported in outputs.
If you want to analyze only specific genomic regions, the input file should have the following 3 columns:
- chr : chromosome
- start : start position of the genomic region of interest (hg19)
- end : end position of the genomic region of interest (hg19)
The order of columns has to be exactly the same as shown above but header could be anything (the first row is ignored). Extra columns will be ignored.
Parameters
Annotation and prioritization depends on several settings, which can be adjusted if desired. The default settings will result in performing naive positional mapping which maps all independent lead SNPs and SNPs in LD to genes up to 10kb apart. It does not include eQTL mapping by default, and it also does not filter on specific functional consequences of SNPs. If for example you are interested in prioritizing genes only when they are indicated by an eQTL that is in LD with a significant lead SNP, or by exonic SNPs, then you need to adjust the parameter settings.
Each of user inputs and parameters have status as described below.
Please make sure all input has non-red status, otherwise the submit button will not be activated.
This is for optional inputs/parameters.
This is the message if everything is fine.
This is the message if the input/parameter is mandatory and not given or invalid input is given.
This is the warning message for the input/parameter. Please check your input settings.
In this section, every parameter that can be adjusted will be described in detail.
1. Input files
| Parameter | Mandatory | Description | Type | Default |
|---|---|---|---|---|
| GWAS summary statistics | Mandatory | Input file of GWAS summary statistics. Plain text file or zipped or gzipped files are acceptable. The maximum file size which can be uploaded is 600Mb. In addition to full results of GWAS summary statistics, subset of results can also be used. e.g. If you would like to look up specific SNPs, you can filter out other SNPs. Please refer to the Input files section for specific file format. | File upload | none |
| Pre-defined lead SNPs | Optional | Optional pre-defined lead SNPs. The file should have 3 columns, rsID, chromosome and position. | File upload | none |
| Identify additional lead SNPs | Optional only when predefined lead SNPs are provided | If this option is CHECKED, FUMA will identify additional independent lead SNPs after defining the LD block for pre-defined lead SNPs. Otherwise, only given lead SNPs and SNPs in LD of them will be used for further annotations. | Check | Checked |
| Pre-defined genetic region | Optional | Optional pre-defined genomic regions. FUMA only looks at provided regions to identify lead SNPs and SNPs in LD of them. If you are only interested in specific regions, this option will increase the speed of process. |
File upload | none |
2. Parameters for lead SNPs and candidate SNPs identification
| Parameter | Mandatory | Description | Type | Default | Direction |
|---|---|---|---|---|---|
| Sample size (N) | Mandatory | The total number of individuals in the GWAS or the number of individuals per SNP.
This is only used for MAGMA to compute the gene-based P-values.
For total sample size, input should be an integer.
When the input file of GWAS summary statistics contains a column of sample size per SNP, the column name can be provided in the second text box. When column name is provided, please make sure that the column only contains integers (no float or scientific notation). If there are any float values, they will be rounded up by FUMA. |
Integer or text | none | Does not affect any candidates |
| Maximum lead SNP P-value (<) | Mandatory | FUMA identifies lead SNPs with P-value less than or equal to this threshold and independent from each other. | numeric | 5e-8 | lower: decrease #lead SNPs. higher: increase #lead SNPs. |
| Maximum GWAS P-value (<) | Mandatory | This is the P-value threshold for candidate SNPs in LD of independent significant SNPs. This will be applied only for GWAS-tagged SNPs as SNPs which do not exist in the GWAS input but are extracted from 1000 genomes reference do not have P-value. | numeric | 0.05 | higher: increase #candidate SNPs. lower: decrease #candidate SNPs. |
| r2 threshold for independent significant SNPs (≥) | Mandatory | The minimum r2 for defining independent significant SNPs, which is used to determine the borders of the genomic risk loci. SNPs with r2 ≥ user defined threshold with any of the detected independent significant SNPs will be included for further annotations and are used fro gene prioritisation. | numeric | 0.6 | higher: decrease #candidate SNPs and increase #independent significant SNPs. lower: increase #candidate SNPs and decrease #independent significant SNPs. |
| 2nd r2 threshold for lead SNPs (≥) | Mandatory | The minimum r2 for defining lead SNPs, which is used for the second clumping (clumping of the independent significant SNPs). Note that when this threshold is same as the first r2 threshold, lead SNPs are identical to independent significant SNPs. | numeric | 0.1 | higher: increase #lead SNPs. lower: decrease #lead SNPs. |
| Reference panel | Mandatory | The reference panel to compute r2 and MAF. Five populations from 1000 genomes Phase 3 and 3 versions of UK Biobank are available. See here for details. | Select | 1000G Phase EUR | - |
| Include variants from reference panel | Mandatory | If Yes, all SNPs in strong LD with any of independent significant SNPs including non-GWAS-tagged SNPs will be included and used for gene mapping. | Yes/No | Yes | - |
| Minimum MAF (≥) | Mandatory | The minimum Minor Allele Frequency to be included in annotation and prioritisation. MAF is based the user selected reference panel. This filter also applies to lead SNPs. If there is any pre-defined lead SNPs with MAF less than this threshold, those SNPs will be skipped. When this value is 0 (by default), SNPs with MAF>0 are considered. | numeric | 0 | higher: decrease #candidate SNPs. lower: increase #candidate SNPs. |
| Maximum distance of LD blocks to merge (≤) | Mandatory | This is the maximum distance between LD blocks of independent significant SNPs to merge into a single genomic locus. When this is set at 0, only physically overlapping LD blocks are merged. Defining genomic loci does not affect identifying which SNPs fulfil selection criteria to be used for annotation and prioritization. It will only result in a different number of reported risk loci, which can be desired when certain loci are partly overlapping or physically very close. | numeric | 250kb | higher: decrease #genomic loci. lower: increase #genomic loci. |
3. Parameters for gene mapping
There are two options for gene mapping; positional and eQTL mappings. By default, positional mapping with maximum distance 10kb is performed. Since parameters in this section largely affect the result of mapped genes, please set them carefully.
3.1 Positional mapping
| Parameter | Mandatory | Description | Type | Default | Direction |
|---|---|---|---|---|---|
| Positional mapping | Optional | Check this option to perform positional mapping. Positional mapping is based on ANNOVAR annotations by specifying the maximum distance between SNPs and genes or based on functional consequences of SNPs on genes. These parameters can be specified in the option below. | Check | Checked | - |
| Distance to genes or functional consequences of SNPs on genes to map | Mandatory if positional mapping is activated. | Positional mapping criterion either map SNPs to genes based on physical distances or functional consequences of SNPs on genes. When maximum distance is provided SNPs are mapped to genes based on the distance given the user defined maximum distance. Alternatively, specific functional consequences of SNPs on genes can be selected which filtered SNPs to map to genes. Note that when functional consequences are selected, all SNPs are locating on the gene body (distance 0) except upstream and downstream SNPs which are up to 1kb apart from TSS or TSE. When the maximum distance is set at > 0kb and < 1kb all upstream and downstream SNPs are included since the actual distance is not provided by ANNOVAR. Therefore, the maximum distance > 0kb and < 1kb is same as the maximum distance 1 kb. For SNPs which are locating on a genomic region where multiple genes are overlapped, ANNOVAR has its own prioritization criteria to report the most deleterious function. For those SNPs, only prioritized annotations are used. |
Integer / Multiple selection | Maximum distance 10 kb | - |
3.2 eQTL mapping
| Parameter | Mandatory | Description | Type | Default | Direction |
|---|---|---|---|---|---|
| eQTL mapping | Optional | Check this option to perform eQTL mapping. eQTL mapping will map SNPs to genes which likely affect expression of those genes up to 1 Mb (cis-eQTL). eQTLs are highly tissue specific and tissue types can be selected in the following option. eQTL mapping can be used together with positional mapping. | Check | Unchecked | - |
| Tissue types | Mandatory if eQTL mapping is CHECKED |
All available tissue types with data sources are shown in the select boxes. From FUMA v1.3.0, GTEx v7 became available but GTEx v6 are kept available. Therefore, when "all" is selected, both GTEx v6 and v7 are used for mapping. For detail of eQTL data resources, please refer to the eQTL section in this tutorial. | Multiple selection | none | - |
| eQTL maximum P-value (≤) | Optional | The P-value threshold of eQTLs.
Two options are available, Use only significant snp-gene pairs or nominal P-value threshold.
When Use only significant snp-gene pairs is checked, only eQTLs with FDR ≤ 0.05 will be used.
Otherwise, defined nominal P-value is used to filter eQTLs.Some of eQTL data source only contained eQTLs with a certain FDR threshold. Please refer to the eQTLs section for details of each data sources. |
Check / Numeric | Checked / 1e-3 | lower: decrease #eQTLs and #mapped genes. higher: increase #eQTLs and #mapped genes. |
3.3 Chromatin interaction mapping
| Parameter | Mandatory | Description | Type | Default | Direction |
|---|---|---|---|---|---|
| chromatin interaction mapping | Optional | Check this option to perform chromatin interaction mapping. | Check | Unchecked | - |
| Builtin chromatin interaction data | Optional | Build in chromatin interaction data can be selected in this option. Details of available build in data are available in the Chromatin interactions section in this tutorial. | Multiple selection | none | - |
| Custom chromatin interaction matrices | Optional | In addition to build in chromatin interaction data, user can upload custom data.
The data should be pre-computed chromatin loops with significance (ideally FDR but another score can be used, see the Chromatin interactions section for details).
The file should be gzipped and named as "(name-of-data).txt.gz". Multiple files can be uploaded.
For each data, user can also provide data type, such as Hi-C, ChIA-PET or C5 which is not mandatory but will be used in the result table and regional plot.
The file format is described in the Chromatin interactions section in this tutorial. Please avoid uploading more than one file with identical file names. In that case, the files are over-written by the last uploaded one. |
File upload (multiple) | none | - |
| FDR threshold (≤) | Mandatory if chromatin interaction mapping is CHECKED |
FDR threshold for significant loops.
The default value is set at 1e-6 which is suggested by Schmitt et al. (2016) This threshold will be applied both build in and user uploaded chromatin loops. |
Numeric | 1e-6 | lower: increase #chromatin interactions and #mapped genes. higher: decrease #chromatin interactions and #mapped genes. |
| Promoter region window | Mandatory if chromatin interaction mapping is CHECKED |
Promoter regions of genes to map in significantly interacting regions. The input format should be "(upstream bp)-(donwstream bp)" from transcription start site (TSS). For example, the default "250-500" means that promoter regions are defined as 250bp upstream and 500bp downstream of the TSS. By the chromatin interaction mapping, genes whose user defined promoter regions are overlapped with the significantly interacting regions will be mapped. Please refer the Chromatin interactions section in this tutorial for details. | text | 250-500 | lower: increase #mapped genes. smaller: decrease #mapped genes. |
| Annotate enhancer/promoter regions (Roadmap 111 epigenomes) | Optional | Predicted enhancer and promoter regions from Roadmap epigenomics project for 111 epigenomes can be annotated to significantly interaction regions. If any epigenome is not selected, enhancer and promoter regions are not annotated. Annotated enhancer/promoter regions can be used to filter SNPs and mapped genes in the next two options. | Multiple selection | none | - |
| Filter SNPs by enhancers | Optional | This option is only available when at least one epigenome is selected in the previous option to annotate enhancer/promoter regions. When this option is checked, SNPs are filtered on such that overlap with one of the annotated enhancer regions for chromatin interaction mapping. Please refer the Chromatin interactions section in this tutorial for details. | Check | Unchecked | - |
| Filter genes by promoters | Optional | This option is only available when at least one epigenome is selected in the previous option to annotate enhancer/promoter regions. When this option is checked, chromatin interaction mapping is only performed for genes whose promoter regions are overlap with one of the annotated promoter regions. Please refer the Chromatin interactions section in this tutorial for details. | Check | Unchecked | - |
3.4 Functional annotation filtering
Positional, eQTL and chromatin interaction mappings have the following options separately, for the filtering of SNPs based on functional annotation. All filters below apply to selected SNPs in LD with independent significant SNPs that are used to prioritize genes and influence the number of SNPs that are mapped to genes, and consequently influence the number of prioritized genes.
| Parameter | Mandatory | Description | Type | Default | Direction |
|---|---|---|---|---|---|
| CADD score | Optional | Check this if you want to perform filtering of SNPs by CADD score. This applies to selected SNPs in LD with independent significant SNPs that are used to prioritize genes. CADD score is the score of deleteriousness of SNPs predicted by 63 functional annotations. 12.37 is the threshold to be deleterious suggested by Amendola et al. (2015). Please refer to https://pubmed.ncbi.nlm.nih.gov/25637381/ for details. | Check | Unchecked | - |
| Minimum CADD score (≥) | Mandatory if CADD score is checked |
The higher the CADD score, the more deleterious. | numeric | 12.37 | higher: less SNPs will be mapped to genes. lower: more SNPs will be mapped to genes. |
| RegulomeDB score | Optional | Check if you want to perform filtering of SNPs by RegulomeDB score. This applies to selected SNPs in LD with independent significant SNPs that are used to prioritize genes. RegulomeDB score is a categorical score representing regulatory functionality of SNPs based on eQTLs and chromatin marks. Please refer to the original publication for details from links. | Check | Unchecked | - |
| Minimum RegulomeDB score (≥) | Mandatory if RegulomeDB score is checked |
RegulomeDB score is a categorical score from 1a to 7) Score 1a means that those SNPs are most likely affecting regulatory elements and 7 means that those SNPs do not have any annotations. SNPs are recorded as NA if they are not present in the database. SNPs with NA will not be included for filtering on RegulomeDB score. | string | 7 | higher: more SNPs will be mapped to genes. lower: less SNPs will be mapped to genes. |
| 15-core chromatin state | Optional | Check if you want to perform filtering of SNPs by chromatin state. This applies to selected SNPs in LD with independent significant SNPs that are used to prioritize genes. The chromatin state represents accessibility of genomic regions (every 200bp) with 15 categorical states predicted by ChromHMM based on 5 chromatin marks for 127 epigenomes. | Check | Unchecked | - |
| 15-core chromatin state tissue/cell types | Mandatory if 15-core chromatin state is checked |
Multiple tissue/cell types can be selected from the list. | Multiple selection | none | - |
| Maximum state of chromatin(≤) | Mandatory if 15-core chromatin state is checked |
The maximum state to filter SNPs. Between 1 and 15. Generally, between 1 and 7 is open state. | numeric | 7 | higher: more SNPs will be mapped to genes. lower: less SNPs will be mapped to genes. |
| Method for 15-core chromatin state filtering | Mandatory if 15-core chromatin state is checked |
When multiple tissue/cell types are selected, either
any (filtered on SNPs which have state above than threshold in any of selected tissue/cell types),
majority (filtered on SNPs which have state above than threshold in majority (≥50%) of selected tissue/cell type), or
all (filtered on SNPs which have state above than threshold in all of selected tissue/cell type).
|
Selection | any | - |
| Annotation datasets | Optional | Additional functional annotations can be annotated to candidate SNPs. All available data are regional based annotation (bed file format). | Multiple selection | none | - |
| Annotation filtering method | Mandatory if any of Annotation datasets is selected. |
By default, SNPs are not filtered by the annotations selected in Annotation datasets.
To filter SNPs based on the selected annotation, select this options from
any (filtered on SNPs which are overlapping with any selected annotations),
majority (filtered on SNPs which are overlapping with majority (≥50%) of selected annotations), or
all (filtered on SNPs which are overlapping with all of selected annotations).
|
Selection | No filtering | - |
4. Gene types
Biotype of genes to map can be selected. Please refer to Ensembl for details of biotypes.
| Parameter | Mandatory | Description | Type | Default |
|---|---|---|---|---|
| Gene type | Mandatory | Gene type to map. This is based on gene_biotype obtained from BioMart of Ensembl. Please see here for details | Multiple selection. | Protein coding genes. |
5. MHC region
The MHC region is often excluded due to its complicated LD structure. Therefore, this option is checked by default. Please uncheck to include MHC region. Note that it doesn't change any results if there is no significant hit in the MHC region.
| Parameter | Mandatory | Description | Type | Default |
|---|---|---|---|---|
| Exclude MHC region | Optional | Check if you want to exclude the MHC region. The default region is defined as between "MOG" and "COL11A2" genes. | Check | Checked |
| Options for excluding MHC region | Optional | MHC region can be excluded only from either annotations or MAGMA gene analysis, or from both by selecting this option. | Select | Only from annotations |
| Extended MHC region | Optional | User specified MHC region to exclude (for extended or shorter region). The input format should be like "25000000-34000000" on hg19. | Text | Null |
6. MAGMA analysis
Starting from FUMA version 1.5.1, user needs to check the magma checkbox to perform MAGMA. MAGMA gene and gene-set analyses are performed for the input summary statistics. Gene expression data sets for MAGMA gene expression analysis can be also selected from here.
| Parameter | Mandatory | Description | Type | Default |
|---|---|---|---|---|
| Perform MAGMA | Optional | CHECK to ENABLE MAGMA analyses. | Check | Unchecked |
| MAGMA gene annotation window | Mandatory when MAGMA is active. |
The window of the genes to assign SNPs (symmetric). e.g. when 5kb is selected, SNPs within 5kb window of a gene (both side) will be assigned to that gene. The option is available from 0, 5, 10, 15, 20kb window. | Select | 0kb from both side of the genes |
| MAGMA gene expression analysis | Mandatory when MAGMA is active. |
Gene expression data sets used for MAGMA gene-property analysis to test positive association between genetic associations and gene expression in a given label. | Select | GTEx v6 |
7. Title of job submission
Title of job submission can be provided at above the "Submit Job" button. This is not mandatory but this would be useful to keep track your jobs.
Outputs of SNP2GENE
Once your job is completed, you will receive an email. Unless an error occurred during the process, the email includes the link to results page (this again requires login). You can also access to the results page from My Job list.
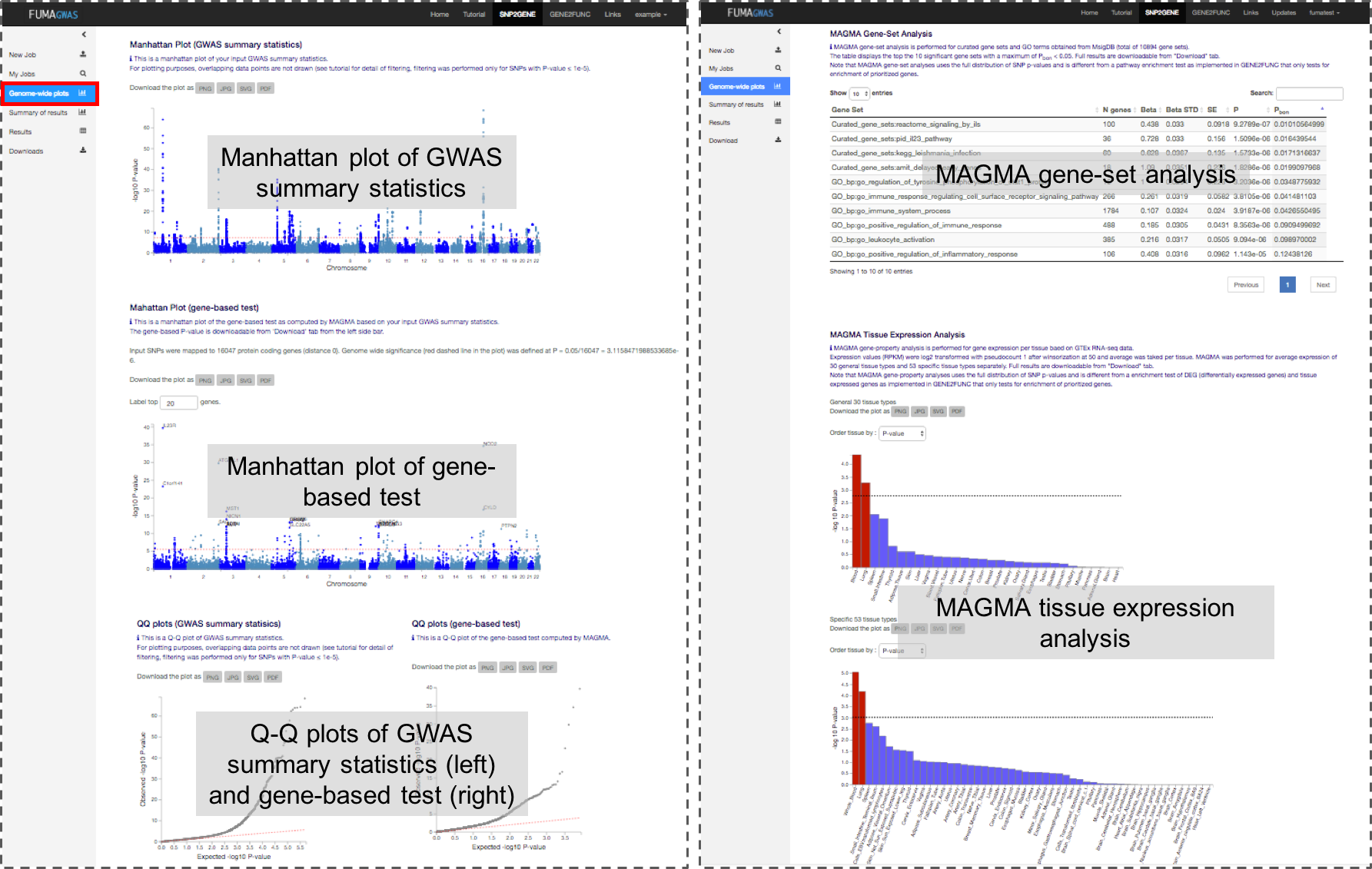
1. Genome-wide plots
This panel displays manhattan plots and Q-Q plots for both GWAS summary statistics (input file) and gene-based association test.
In addition MAGMA based gene-set P-values are provided.
Note that MAGMA gene-set analysis uses the full distribution of SNP p-values and is different from pathway enrichment test that only test for enrichment of low P-values, or enrichment of prioritized genes.
Images are downloadable in several formats, and underlying results can be downloaded in Table format from the download tab.
Plots for GWAS summary statistics
For plotting purposes, overlapping data points are filtered out based on the following criteria.
- Manhattan plot: Overlapping data points (SNPs) were filtered out such that there is only one data point per pixel, but only when the average data points per pixel (x-axis) across y-axis is above 1. For each pixel, the plotted data point was randomly selected. SNPs with P-value ≥ 1e-5 are removed.
Plots for gene-based test (MAGMA)
Gene analysis was performed by using MAGMA (v1.10 with default setting.
SNPs were assigned to the genes obtained from Ensembl build 85 (only protein-coding genes).
Genome-wide significance (red dashed line) was set at 0.05 / (the number of tested genes).
Genes whose P-value reached the genome-wide significance can be labeled in the manhattan plot.
The number of genes to label can be controlled by typing the number at the left upper side of the plot.
MAGMA results are available from the download panel.
When the option is selected to exclude MHC region from MAGMA gene analysis, the results of MAGMA does not include MHC region,
therefore manhattan plot also does not display genes in MHC region.
MAGMA Gene-Set Analysis
Using the result of gene analysis (gene level p-value), (competitive) gene-set analysis is performed with default parameters with MAGMA v1.10.
Gene sets were obtained from Msigdb v7.0 for "Curated gene sets" and "GO terms".
MAGMA Tissue Expression Analysis (FUMA v1.1.0)
To test the (positive) relationship between highly expressed genes in a specific tissue and genetic associations, gene-property analysis is performed using average expression of genes per tissue type as a gene covariate.
Gene expression values are log2 transformed average RPKM per tissue type after winsorized at 50 based on GTEx RNA-seq data. Tissue expression analysis is performed for 30 general tissue types and 53 specific tissue types separately.
MAGMA was performed using the result of gene analysis (gene-based P-value) and tested for one side (greater) with conditioning on average expression across all tissue types.
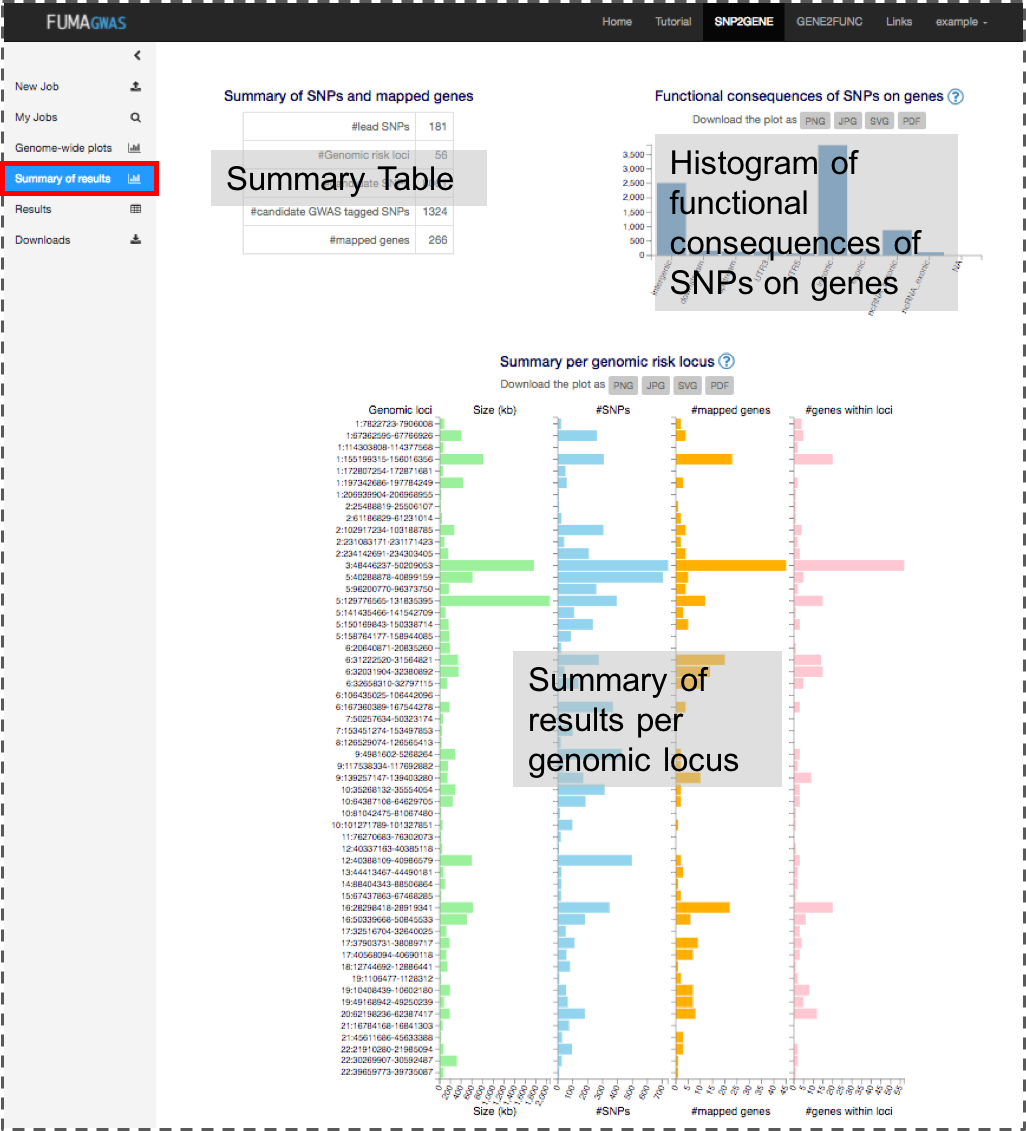
2. Summary of results
This panel shows a general summary of the results based on your GWAS input. Images are downloadable in several formats.
- Summary of SNPs and mapped genes
- #Genomic risk loci: The number of genomic risk loci defined from independent significant SNPs by merging LD blocks if they are less apart than the user defined distance. A genomic risk locus can contain multiple lead SNPs and/or independent significant SNPs.
- #lead SNPs: The number of lead SNPs identified from independent significant SNPs which are independent each other at r2 0.1.
- #independent significant SNPs: The number of independent significant SNPs which reached the user defined genome-wide significant P-value and are independent each other at the user defined r2
- #candidate SNPs: The number of candidate SNPs which are in LD (given r2) of one of the independent significant SNPs. This includes non-GWAS tagged SNPs which are extracted from the 1000 genomes reference panel. When SNPs are filtered based on functional annotation for gene mapping, this number refers to the number of SNPs before the functional filtering.
- #candidate GWAS tagged SNPs: The number of candidate SNPs (described above) which are tagged in GWAS (exists in your input file).
- #mapped genes: The number of genes mapped based on the user-defined parameters.
- Positional annotation of candidate SNPs
This is a histogram of the number of SNPs per functional consequences on genes. When SNPs have more than one (different) annotations, they are counted for each annotation. SNPs assigned NA; this may be because alleles do not match with the fasta files of ANNOVAR Ensembl genes. - Summary per genomic locus
This histogram displays the size of genomic risk loci, the number of candidate SNPs, the number of prioritized genes and the number of genes physically locating within the genomic locus.
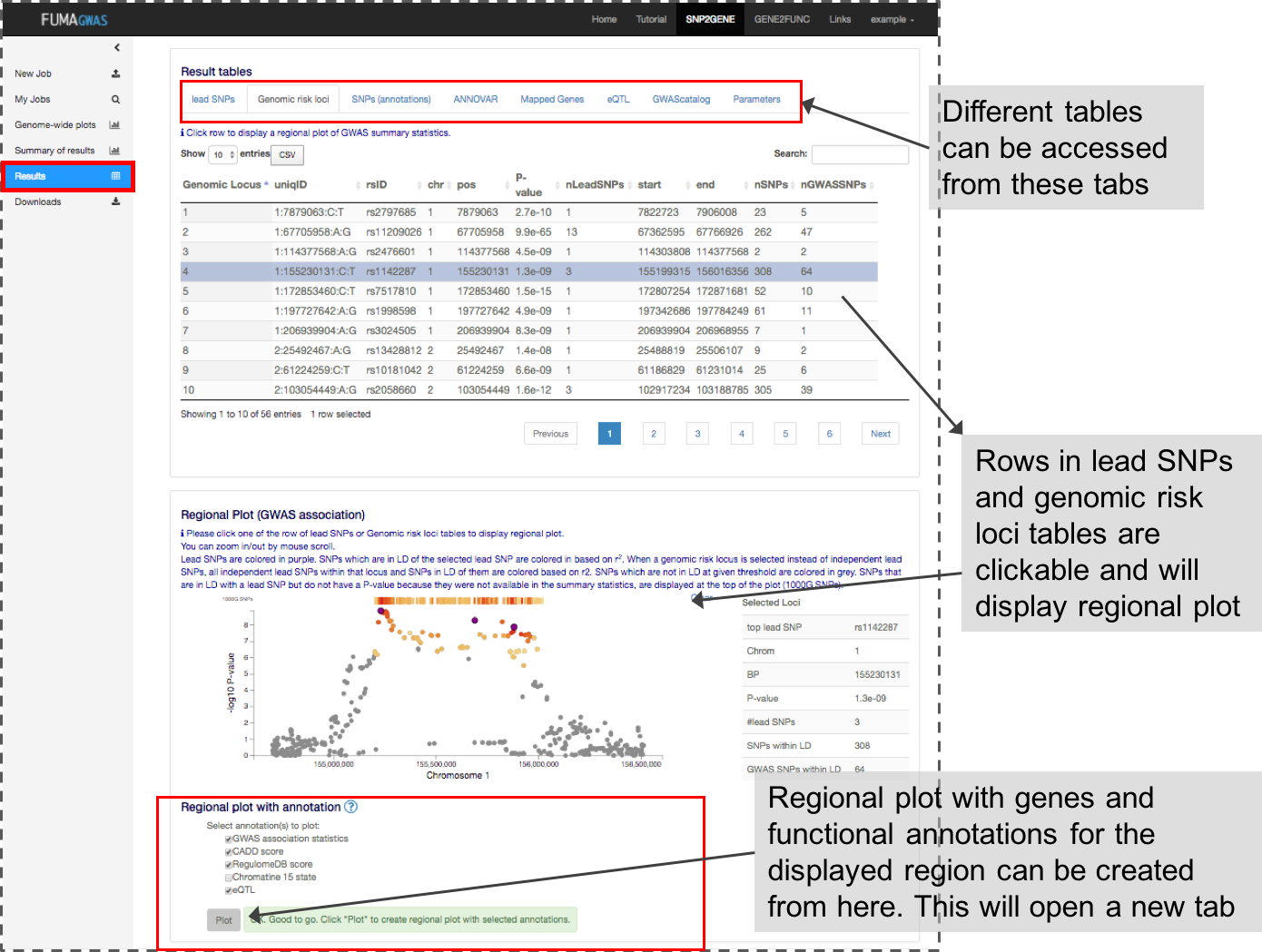
3. Result tables
This panel contains multiple tables of the results. Each column is described in Table columns.
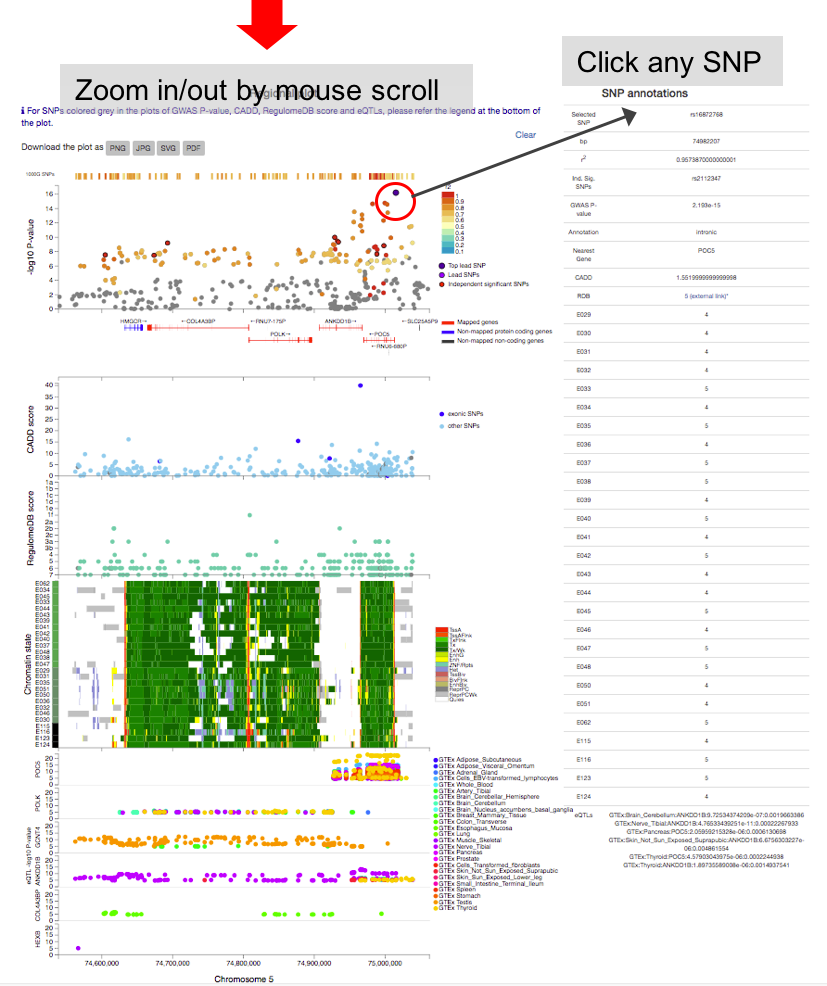
By clicking one of the rows of tables of genomic risk loci, lead SNPs or independent significant SNPs, FUMA will create regional plots of candidate SNPs (GWAS P-value). Optionally, regional plots with genes and functional annotations can be created from the panel at the bottom of the page.
Regional plots can be created with the following optional annotations:- GWAS association statistics: input P-value
- CADD score
- RegulomeDB score
- 15-core chromatin state: tissue/cell types have to be selected.
- eQTLs: This option is only available when eQTL mapping is performed. eQTLs are plotted per gene and colored per tissue types.
- chromatin interactions: This option is only available when chromatin mapping is performed. Interactions are plotted per data set.
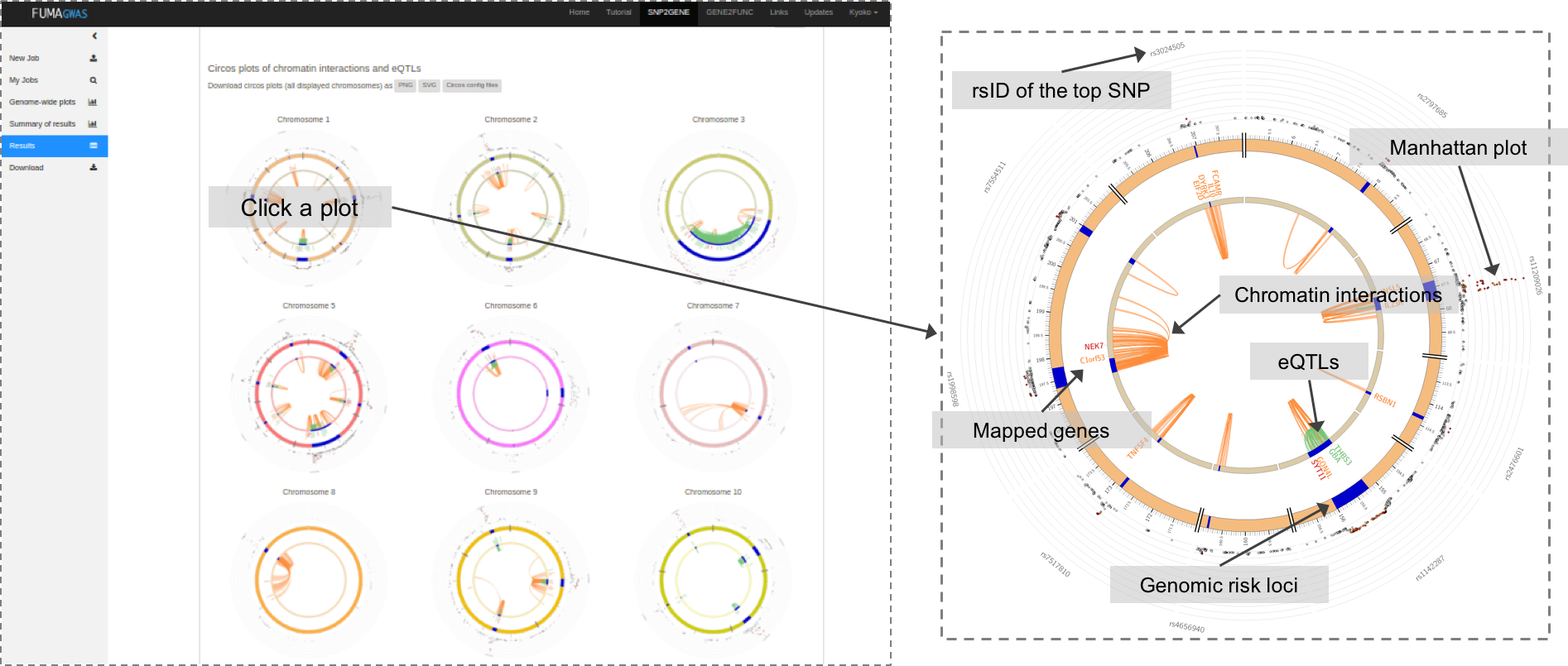
When chromatin interaction mapping is performed, circos plots are created for each chromosome that contains at least one risk locus.
The circos plots are displayed in the panel where the chromatin interaction tables are displayed.
Each plot is clickable and opens in a new tab showing a larger plot.
PNG, SVG and circos config files are downloadable.
All chromatin interactions overlapping with any of risk loci (including interactions that do not map to genes) will be shown in the circos plot.
The specific layers and color-coding of the circos plot is described below.
- Manhattan plot: The most outer layer. Only SNPs with P < 0.05 are displayed.
SNPs in genomic risk loci are color-coded as a function of their maximum r2 to the one of the independent significant SNPs in the locus, as follows:
red (r2 > 0.8), orange (r2 > 0.6), green (r2 > 0.4) and blue (r2 > 0.2). SNPs that are not in LD with any of the independent significant SNPs (with r2 ≤ 0.2) are grey.
The rsID of the top SNPs in each risk locus are displayed in the most outer layer. Y-axis are raned between 0 to the maximum -log10(P-value) of the SNPs. - Chromosome ring: The second layer. Genomic risk loci are highlighted in blue.
- Mapped genes by chromatin interactions or eQTLs: Only mapped genes by either chromatin interaction and/or eQTLs (conditional on user defined parameters) are displayed. If the gene is mapped only by chromatin interactions or only by eQTLs, it is colored orange or green, respectively. When the gene is mapped by both, it is colored red.
- Chromosome ring: The third layer. This is the same as second layer but without coordinates to make it easy to align position of genes with genomic coordinate.
- Chromatin interaction links: Links colored orange are chromatin interactions. Since v1.2.7, only the interactions used for mapping based on user defined parameters are displayed.
- eQTL links: Links colored green are eQTLs. Since v1.2.7, only the eQTLs used for mapping based on user defined parameters are displayed.
4. Downloads
All results are downloadable as text file.
Columns are described in Table columns.
README file is also included in a zip file.
When the SNP table is selected to downloaded, ld.txt will be also included in the zip file.
This file contains the r2 values computed from selected reference panel for all pairs of one of the independent significant SNPs and all other SNPs within the LD.
Table Columns
Genomic risk loci
- Genomic locus : Index of genomic rick loci.
- uniqID : Unique ID of SNPs consisting of chr:position:allele1:allele2 where alleles are alphabetically ordered.
- rsID : rsID of the top lead SNP based on dbSNP build 146.
- chr : chromosome of top lead SNP
- pos : position of top lead SNP on hg19
- P-value : P-value of top lead SNP (from the input file).
- start : Start position of the locus
- end : End position of the locus
- nSNPs : The number of unique candidate SNPs in the genomic locus, including non-GWAS-tagged SNPs (which are available in the user selected reference panel). Candidate SNPs are all SNPs that are in LD (give user-defined r2) with any of independent significant SNPs and either have a P-value below the user defined threshold or are only available in 1000G.
- nGWASSNPs : The number of unique GWAS-tagged candidate SNPs in the genomic locus which is available in the GWAS summary statistics input file. This is a subset of "nSNPs".
- nIndSigSNPs : The number of the independent (at user defined r2) significant SNPs in the genomic locus.
- IndSigSNPs : rsID of the independent significant SNPs in the genomic locus.
- nLeadSNPs : The number of lead SNPs in the genomic locus. Lead SNPs are subset of independent significant SNPs at r2 0.1.
- LeadSNPs : rsID of lead SNPs in the genomic locus.
lead SNPs
- No : Index of lead SNPs
- Genomic Locus : Index of assigned genomic locus matched with "Genomic risk loci" table. Multiple lead SNPs can be assigned to the same genomic locus.
- uniqID : Unique ID of SNPs consisting of chr:position:allele1:allele2 where alleles are alphabetically ordered.
- rsID : rsID based on dbSNP build 146.
- chr : chromosome
- pos : position on hg19
- P-value : P-value (from the input file).
- nIndSigSNPs : Number of independent significant SNPs which are in LD with the lead SNP at r2 0.1.
- IndSigSNPs : rsID of independent significant SNPs which are in LD with the lead SNP at r2 0.1.
independent significant SNPs (Independent significant SNPs)
All independent lead SNPs identified by FUMA.
- No : Index of independent significant SNPs
- Genomic Locus : Index of assigned genomic locus matched with "Genomic risk loci" table. Multiple independent lead SNPs can be assigned to the same genomic locus.
- uniqID : Unique ID of SNPs consisting of chr:position:allele1:allele2 where alleles are alphabetically ordered.
- rsID : rsID based on dbSNP build 146.
- chr : chromosome
- pos : position on hg19
- P-value : P-value (from the input file).
- nSNPs : The number of SNPs in LD with the lead SNP given r2, including non-GWAS-tagged SNPs (which are extracted from 1000G).
- nGWASSNPs : The number of GWAS-tagged SNPs in LD with the lead SNP given r2. This is a subset of "nSNPs".
SNPs
All candidate SNPs (SNPs which are in LD of any independent lead SNPs) with annotations. Note that depending on your mapping criterion, not all candidate SNPs displaying in this table are mapped to genes.
- uniqID : Unique ID of SNPs consisting of chr:position:allele1:allele2 where alleles are alphabetically ordered.
- rsID : rsID based on dbSNP build 146.
- chr : chromosome
- pos : position on hg19
- effect_allele : Effect/risk allele if it is provided in the input GWAS summary statistics file. If not, this is the alternative (minor) allele in 1000G.
- non_effect_allele : Non-effect/non-risk allele if it is provided in the input GWAS summary statistics file. If not, this is the reference (major) allele in 1000G.
- MAF : Minor allele frequency computed based on 1000G.
- gwasP : P-value provided in the input GWAS summary statistics file. Non-GWAS tagged SNPs (which do not exist in input file but are extracted from the reference panel) have "NA" instead.
- or : Odds ratio provided in the input GWAS summary statistics file if available. Non-GWAS tagged SNPs (which do not exist in input file but are extracted from the reference panel) have "NA" instead.
- beta : Beta provided in the input GWAS summary statistics file if available. Non-GWAS tagged SNPs (which do not exist in input file but are extracted from the reference panel) have "NA" instead.
- se : Standard error provided in the input GWAS summary statistics file if available. Non-GWAS tagged SNPs (which do not exist in input file but are extracted from the reference panel) have "NA" instead.
- r2 : The maximum r2 of the SNP with one of the independent significant SNPs.
- IndSigSNP : rsID of the independent significant SNP which has the maximum r2 with the SNP.
- Genomic locus : Index of the genomic risk loci matching with "Genomic risk loci" table.
- nearestGene : The nearest Gene of the SNP based on ANNOVAR annotations. Note that ANNOVAR annotates "consequence" function by prioritizing the most deleterious annotation for SNPs which are locating a genomic region where multiple genes are obverlapped. Genes are ecoded in symbol, if it is available otherwise Ensembl ID. Genes include all transcripts from Ensembl gene build 85 including non-protein coding genes and RNAs.
- dist : Distance to the nearest gene. SNPs which are locating in the gene body or 1kb up- or down-stream of TSS or TES have 0.
- func : Functional consequence of the SNP on the gene obtained from ANNOVAR. For exonic SNPs, detailed annotation (e.g. non-synonymous, stop gain and so on) is available in the ANNOVAR table (annov.txt).
- CADD : CADD score which is computed based on 63 annotations. The higher the score, the more deleterious the SNP is. 12.37 is the suggested threshold by Kicher et al (2014).
- RDB : RegulomeDB score which is a categorical score (from 1a to 7). 1a is the highest score for SNPs with the most biological evidence to be a regulatory element.
- minChrState : The minimum 15-core chromatin state across 127 tissue/cell type.
- commonChrState : The most common 15-core chromatin state across 127 tissue/cell types.
- posMapFilt : Whether the SNP was used for eQTL mapping or not. 1 is used, otherwise 0. When eqtl mapping is not performed, all SNPs have 0.
Complete annotations of 15-core chromatin state (for every 127 epigenomes) are available in the "annot.txt" from download.
ANNOVAR
Since one SNP can be annotated to multiple positional information, the table of ANNOVAR output is separated from SNPs table. This table contains unique SNP-annotation combinations.
- uniqID : Unique ID of SNPs consisting of chr:position:allele1:allele2 where alleles are alphabetically ordered.
- chr : chromosome
- pos : position on hg19
- Gene : ENSG ID
- Symbol : Gene Symbol
- Distance : Distance to the gene
- Function : Functional consequence on the gene
- Exonic function : Functional annotation of exonic SNPs
- Exon : Index of exon
Mapped genes
The genes which are mapped by SNPs in the SNPs table based on user-defined mapping parameters. Columns with posMap, eqtlMap or ciMap in the parentheses are only available when positional, eQTL or chromatin interaction mapping is performed, respectively.
- Gene : ENSG ID
- Symbol : Gene Symbol
- entrezID : entrez ID
- Genomic locus : Index of genomic loci where mapped SNPs are from. This could contain more than one interval in the case that eQTLs are mapped to genes from distinct genomic risk loci.
- chr : chromosome
- start : Starting position of the gene
- end : Ending position of the gene
- strand : Strand of gene
- status : Status of gene from Ensembl
- type : Gene biotype from Ensembl
- HUGO : HUGO (HGNC) gene symbol
- pLI : pLI score from ExAC database. The probability of being loss-of-function intolerant. The higher the score is, the more intolerant to loss-of-function mutations the gene is.
- ncRVIS : Non-coding residual variation intolerance score. The higher the score is, the more intolerant to noncoding variants the gene is.
- posMapSNPs (posMap): The number of SNPs mapped to gene based on positional mapping (after functional filtering if parameters are given).
- posMapMaxCADD (posMap): The maximum CADD score of mapped SNPs by positional mapping.
- eqtlMapSNPs (eqtlMap): The number of SNPs mapped to the gene based on eQTL mapping.
- eqtlMapminP (eqtlMap): The minimum eQTL P-value of mapped SNPs.
- eqtlMapminQ (eqtlMap): The minimum eQTL FDR of mapped SNPs.
- eqtlMapts (eqtlMap): Tissue types of mapped eQTL SNPs.
- eqtlDirection (eqtlMap): Consecutive direction of mapped eQTL SNPs after aligning risk increasing alleles in GWAS and tested alleles in eQTL data source.
- ciMap (ciMap): "Yes" if the gene is mapped by chromatin interaction mapping.
- ciMapts (ciMap): Tissue/cell types of mapped chromatin interactions.
- minGwasP : The minimum P-value of mapped SNPs.
- IndSigSNPs : rsID of the all independent significant SNPs of mapped SNPs.
eQTL
This table is only shown when eQTL mapping is performed. The table contains unique pairs of SNP-gene-tissue, therefore, a SNP could appear multiple times.
- uniqID : Unique ID of SNPs consisting of chr:position:allele1:allele2 where alleles are alphabetically ordered.
- chr : chromosome
- pos : position on hg19
- DB : Data source of eQTLs. Currently GTEx, BloodeQTL, BIOS and BRAINEAC are available. Please refer to the eQTL section for details.
- tissue : Tissue type
- Gene : ENSG ID
- Symbol : Gene symbol
- P-value : P-value of eQTLs
- FDR : FDR of eQTLs. Note that the method to compute FDR differs between data sources. Please refer to the eQTL section for details.
- signed_stats : Signed statistics, the actual value depends on the data source. Please refer to the eQTL sectuib fir details.
- RiskIncAllele : Risk increasing allele obtained from the input GWAS summary statistics.
"NA" if signed effect is not provided in the input file. SNPs which are not in the input GWAS but included from reference panel are also encoded as "NA". - alignedDirection : The direction of effect to gene expression after aligning risk increasing allele of GWAS and tested allele of eQTLs.
Chromatin interaction (Chromatin interactions tab)
This file is only available when chromatin interaction mapping is performed. The file contains significant interactions of user defined data or user uploaded data filtered.
- GenomicLocus : Index of genomic loci where the significant interaction is overlapped.
- region1 : One end of significant chromatin interaction which overlap with at least one candidate SNPs in one of the genomic risk loci.
- region2 : The other end of significant chromatin interaction. This region could be located outside the risk loci.
- FDR : FDR of interaction.
- type : Type of chromatin interaction data, e.g. Hi-C or ChIA-PET
- DB : The name of data source.
- tissue/cell : Tissue or cell type of the interaction.
- intra/inter : Intra- or Inter-chromosomal interaction.
- SNPs : rsID of candidate SNPs which are overlapping with the region 1.
- genes : ENSG ID of genes whose promoter regions are overlapped with region 2.
SNPs and overlapped regulatory elements in region 1 (Chromatin interaction tab)
This file is only available when chromatin interaction mapping is performed. The file contains candidate SNPs which overlap with one end (region 1) of significant chromatin interaction and enhancer regions of user selected epigenomes. If any epigenome was selected, this file is empty.
- uniqID : Unique ID of SNPs consists of chr:position:allele1:allele2 where alleles are alphabetically ordered.
- rsID : rsID based on dbSNP build 146
- chr : chromosome
- pos : position on hg19
- reg_region : Predicted enhancer or dyadic regions
- type : enh for enhancer and dyadic for dyadic enhancer/promoter regions
- tissue/cell : EID of 111 Roadmap epigenomes
Regulatory elements and genes in region 2 (Chromatin interaction tab)
This file is only available when chromatin interaction mapping is performed. The file contains promoter regions of user selected epigenomes (if selected any) and genes whose promoter regions are overlapped. If any epigenome was selected, this file is empty.
- region2 : region 2 in the chromatin interaction table
- reg_region : Predicted promoter or dyadic regions
- type : prom for promoter and dyadic for dyadic enhancer/promoter regions
- tissue/cell : EID of 111 Roadmap epigenomes
- genes : genes whose promoter regions are overlapped with region2
GWAScatalog
List of SNPs reported in GWAScatalog which are candidate SNPs of your GWAS summary statistics.
The table does not show all columns available. The complete table is available by downloading.- Genomic locus : Index of genomic risk loci.
- IndSigSNP : One of the independent significant SNPs of the SNP in GWAScatalog.
- chr : chromosome
- bp : position on hg19
- snp : rsID of reported SNP in GWAS catalog
- PMID : PubMed ID
- Trait : The trait reported in GWAScatalog
- FirthAuth : First author reported in GWAScatalog
- Date : Date added in GWAScatalog
- P-value : Reported P-value
Parameters
The table of input parameters. The downloadable file is a config file with INI format.
-
[jobinfo]
- created_at : Date of job created
- title : Job title [inputfiles]
- gwasfile : File name of GWAS summary statistics
- leadSNPsfile : File name of pre-defined lead SNPs if provided.
- addleadSNPs : 1 if option is checked, 0 otherwise. If pre-defined lead SNPs are not provided, it is always 1.
- regionsfile : File name of pre-defined genetic regions if provided.
- **col : The column names of input GWAS summary statistics file if provided. [params]
- N : Sample size of GWAS
- exMHC : 1 to exclude MHC region, 0 otherwise
- extMHC : user defined MHC region if provided, NA otherwise
- genetype : All selected gene type.
- leadP : the maximum threshold of P-value to be lead SNP
- r2 : the minimum threshold for SNPs to be in LD of the lead SNPs
- gwasP : the maximum threshold of P-value to be candidate SNP
- pop : The population of reference panel
- MAF : the minimum minor allele frequency based on 1000 genome reference of given population
- Incl1KGSNPs : 1 to include non-GWAS-tagged SNPs from reference panel, 0 otherwise
- mergeDist : The maximum distance between LD blocks to merge into interval [posMap]
- posMap : 1 to perform positional mapping, 0 otherwise
- posMapWindowSize : If provided, this distance is used as the maximum distance between SNPs to genes. Otherwise "NA".
- posMapAnnot : Functional consequences of SNPs on genes to map.
- posMapCADDth : The minimum CADD score for SNP filtering
- posMapRDBth : The minimum RegulomeDB score for SNP filtering
- posMapChr15 : Select tissue/cell types, NA otherwise
- posMapChr15Max : The maximum 15-core chromatin state
- posMapChr15Meth : The method of chromatin state filtering [eqtlMap]
- eqtlMap : 1 to perform eQTL mapping, 0 otherwise
- eqtlMaptss : Selected tissue typed for eQTL mapping
- eqtlMapSig : 1 to use only significant snp-gene pairs, 0 otherwise
- eqtlMapP : The P-value threshold for eQTLs if
eqtlMap significant onlyis not selected. - eqtlMapCADDth : The minimum CADD score for SNP filtering
- eqtlMapRDBth : The minimum RegulomeDB score for SNP filtering
- eqtlMapChr15 : Select tissue/cell types, NA otherwise
- eqtlMapChr15Max : The maximum 15-core chromatin state
- eqtlMapChr15Meth : The method of chromatin state filtering [ciMap]
- ciMap : 1 to perform chromatin interaction mapping, 0 otherwise
- ciMapBuiltin : Selected builtin chromatin interaction data
- ciMapFileN : The number of uploaded chromatin interaction matrices
- ciMapFiles: File names of uploaded chromatin interaction matrices
- ciMapFDR : The FDR threshold of chromatin interactions
- ciMapPromWindow : Window of the promoter regions from TSS. 250-500 means, 250bp up- and 500bp down-stream of TSS region is defined as promoter.
- ciMapRoadmap : Select epigenome ID of roadmap epigenomes for annotation of promoter/enhancer regions
- ciMapEnhFilt : 1 to filter SNPs on such that are overlapped with annotated enhancer regions of selected epigenomes, 0 otherwise
- ciMapPromFilt: 1 to filter mapped genes on such that whose promoter regions are overlapped with annotated promoter regions of selected epigenomes, 0 otherwise
- ciMapCADDth : The minimum CADD score for SNP filtering
- ciMapRDBth : The minimum RegulomeDB score for SNP filtering
- ciMapChr15 : Select tissue/cell types, NA otherwise
- ciMapChr15Max : The maximum 15-core chromatin state
- ciMapChr15Meth : The method of chromatin state filtering
Redo gene mapping for existing jobs
From FUMA v1.3.0, gene mapping can be re-performed for existing job with a different parameter setting. This allows users to tune gene mapping parameters without performing entire process again, by duplicating the selected job, which reduce a large amount of time.1. Select a jobID to duplicate
At the top of the page, users can select a jobID of existing job on the account. Note that only jobs which are succeeded are selectable. This is only available for users who already have SNP2GENE jobs.2. Modify parameters
Once a jobID is selected, the previous parameters are automatically loaded. Modify parameters before submitting, otherwise the results will be same as the selected job. For chromatin interaction mapping, user custom files need to be re-uploaded.Users are allowed to provide new title and suffix "_copied_(jobID)" will be automatically added to the title.
Users are only allowed to modify gene mapping parameters. Other parameters such as P-value or r2 threshold for defining independent significant SNPs cannot be changed.
3. Submit
User can submit the job by clicking the button at the bottom of the page. After submission, the process is same as submitting a new SNP2GENE job, you will get an email once the process is done and results are accessible from your job list table.Reference panel
To define independent significant SNPs, lead SNPs and genomic risk loci, FUMA uses reference panels. In this section, each reference panel is described details.From FUMA v1.3.5, multi allelic SNPs are all included.
1. 1000 Genome Phase3
Genotype data for chromosome 1-22 and X was downloaded from ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/.Multi allelic SNPs were first split into separate columns using vcfmulti2oneallele.jar from JVARKIT (http://lindenb.github.io/jvarkit/). VCF files were then converted to PLINK bfile (PLINK v1.9). Any CNVs were removed, while any indels were kept. Unique ID (consists of chr:position:allele1:allele2 where alleles were alphabetically ordered) was assigned to each SNP and duplicated SNPs (with identical unique ID) were excluded. Genotype data were split into 5 (super) populations based on panel file (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/integrated_call_samples_v3.20130502.ALL.panel) using PLINK.
MAF and pairwise LD were computed by PLINK (--r2 --ld-window 99999 --ld-window-r2 0.05) for each population and all samples together (ALL), and SNPs with MAF=0 were excluded for each population.
Reference panel ALL covers most number of SNPs. To avoid missing SNPs from FUMA annotations, reference panel ALL might be preferred. However, the LD is not population specific and need caution for the definition of independent significant SNPs and lead SNPs.
Number of samples and SNPs in the reference panels (click on a row to download the corresponding variant file):
| Population | Sample size | Number of SNPs | Download size |
|---|---|---|---|
| ALL | 2,504 | 84,853,668 |  870M 870M |
| AFR | 661 | 43,676,209 | 461M |
| AMR | 347 | 29,501,504 | 305M |
| EAS | 504 | 24,507,348 | 254M |
| EUR | 503 | 25,063,419 | 260M |
| SAS | 489 | 27,691,316 | 287M |
2. UK Biobank release 2b
Genotype data was obtained under application ID 16406. The reference panel is based on genotype data released in May 2018 (including SNPs imputed UK10K/1000G). Two reference panels were created; white British and European subjects. For white British, 10,000 unrelated individuals were randomly selected. For European, each individuals were first assigned to one of the 5 1000G populations based on the minimum Mahalanobis distance. Then randomly selected 10,000 unrelated EUR individuals were used.SNPs were filtered on INFO score > 0.9. MAF and pairwise LD were computed by PLINK (--r2 --ld-window 99999 --ld-window-r2 0.05) and SNPs with MAF=0 were excluded.
In both reference panels, 16,972,700 SNPs are available.
ANNOVAR enrichment test
Enrichment of functional consequences of SNPs are tested agains the user selected reference panel. All SNPs that are in LD with one of the independent significant SNPs are annotated by ANNOVAR. SNPs can be annotated to multiple annotations, and those SNPs are counted twice. If SNPs have same annotations assigned to more than one gene, those SNPs are counted once (i.e. only unique combinations of SNP-annotation are counted). There might SNPs that are not annotated by ANNOVAR which are not included in the enrichment test. Thus, sum of counts across annotation ("count" column in "annov.stats.txt" file) is not necessary the same as the number of SNPs in "snps.txt" file. Same applies to the counts for reference panel (sum of "ref.count" is not necessary the same as the number of SNPs mentioned in the "Reference panel" section of this tutorial).Enrichment value is computed as (proportion of SNPs with an annotation) /(proportion of SNPs with an annotation relative to all available SNPs in the reference panel). Fisher's exact test (two side) is performed for each annotation as below.
# count: a vector of the number of SNPs for each annotation
# ref.count: a vector of the number of SNPs for each annotation for all SNPs in the reference panel
N = sum(ref.count)
n = sum(count)
# to compute P-value of the first annotation with R, for example
fisher.test(matrix(c(count[1], n-count[1], ref.count[1]-count[1], N-n-ref.count[1]), ncol=2))
MAGMA analyses
FUMA performs MAGMA gene analysis, gene-set analysis and gene-property analysis. In this section, reference panels and gene expression data sets are described detils.Reference panel
1. 1000 Genome Phase3
Same as described in Reference panel section.2. UK Biobank release2
Same as described in Reference panel section, except further 1,000 individuals were randomly selected to reduce runtime of MAGMA (it takes >3 hours with 10,000 individuals).Gene analysis
The command FUMA uses is the following.
magma --bfile [path to the selected reference panel] \
--pval [magma input file] ncol=3 (or N=[total sample size]) \
--gene-annot [path to the annotation file with use selected window size] \
--out [output file]
Gene set analyses
For FUMA ≤ v1.3.0, 10894 gene sets (curated gene sets: 4728, GO terms: 6166) from MsigdB v5.2 are used.
For FUMA ≥ v1.3.1, 10655 gene sets (curated gene sets: 4738, GO terms: 5917) from MsigDB v6.1 are used.
For FUMA ≥ v1.3.4, 10678 gene sets (curated gene sets: 4761, GO terms: 5917) from MsigDB v6.2 are used.
For FUMA ≥ v1.5.5, 15496 gene sets (curated gene sets: 5500, GO terms: 9996) from MsigDB v7.0 are used.
For FUMA ≥ v1.5.6, 17023 gene sets (curated gene sets: 6494, GO terms: 10529) from MsigDB v2023.1Hs are used.
Bonferroni correction was performed for the all tested gene sets.
To customise, you can download the output file and select a specific gene sets.
The MSigDB v7.0 gene-set file used in FUMA from version 1.3.5d to 1.5.5 can be downloaded here:
22M
24M
Gene set analysis is performed by the following command.
magma --gene-results [path to]/magma.genes.raw \
--set-annot [path to gene set file] \
--out [output file]
Gene property analysis for tissue specificity
MAGMA gene-property analysis is run with the following command,
magma --gene-results [input file name].genes.raw \
--gene-covar [file name of selected RNA-seq data set] \
--model direction-covar=greater condition-hide=Average \
--out [output file name]
Gene expression data sets
1. GTEx v6
Data source
RNAseq data set was downloaded from http://www.gtexportal.org/home/datasets.
Gene level RPKM was used (GTEx_Analysis_v6_RNA-seq_RNA-SeQCv1.1.8_gene_rpkm.gct.gz).
Pre-process
Primary gene ID was Ensemble ID.
In total, 8,555 samples were available.
From 56,318 annotated genes, genes were filtered on such that average RPKM per tissue is >1 in at least on of the 53 tissues.
This resulted in 28,577 genes.
RPKM was winsorized at 50 (replaced RPKM>50 with 50).
Then average of log transformed RPKM with pseudocount 1 (log2(RPKM+1)) per tissue (for either 53 detail or 30 general tissues)
was used as the covariates conditioning on the average across all the tissues.
2. GTEx v7
Data source
RNAseq data set was downloaded from http://www.gtexportal.org/home/datasets.
Gene level TPM was used (GTEx_Analysis_2016-01-15_v7_RNASeQCv1.1.8_gene_rpm.gct.gz).
Pre-process
Primary gene ID was Ensemble ID.
In total, 11,688 samples were available.
From 56,203 annotated genes, genes were filtered on such that average TPM per tissue is >1 in at least on of the 53 tissues.
This resulted in 32,335 genes.
TPM was winsorized at 50 (replaced TPM>50 with 50).
Then average of log transformed TPM with pseudocount 1 (log2(TPM+1)) per tissue (for either 53 detail or 30 general tissues)
was used as the covariates conditioning on the average across all the tissues.
3. BrainSpan
Data source
RNAseq data set was downloaded from http://www.brainspan.org/static/download.
Gene level RPKM was used (genes_matrix_csv.zip).
Pre-process
Primary gene ID was Ensemble ID.
In total, 524 samples were available.
General developmental stages were annotated for each sample based on the age.
We used 11 developmental stages and 29 ages as the label.
For the label of age, we excluded age groups with <3 samples (25 pcw and 35 pcw).
From 52,376 annotated genes, genes were filtered on such that average RPKM per label is >1 in at least one of the either developmental stage or age.
This resulted in 19,601 and 21,001 genes for developmental stages and age groups, respectively.
RPKM was winsorized at 50 (replaced RPKM>50 with 50).
Then average of log transformed RPKM with pseudocount 1 (log2(RPKM+1)) per label (for either 11 developmental stages or 29 age groups)
was used as the covariates conditioning on the average across all the labels.
Risk loci and lead SNPs
In this section, "Genomic risk loci", "lead SNPs" and "Independent significant SNPs (Ind. sig. SNPs)" are explained in more detail.From FUMA v1.3.5, r2 threshold for the second clumping can be provided by users.
1. Independent significant SNPs (Ind. sig. SNPs)
Ind. sig. SNPs are defined as SNPs that have a P-value ≤ the user define threshold for genome-wide significance (5e-8 by default) and are independent from each other at the user defined r2 (0.6 by default). Therefore, ind. sig. SNPs are essentially the same as SNPs that are contained after clumping GWAS tagged SNPs at the same P-value and r2. Ind. sig. SNPs are used to select candidate SNPs that are in LD with the ind. sig. SNPs.The candidate SNPs (and ind. sig. SNPs) are used for gene prioritization.
Relaxing the threshold for the genome-wide significant P-value results in an increased number of ind. sig. SNPs. When you would like to identify ind. sig. SNPs in genomic loci which do not reach the commonly adopted genome-wide significance level of 5e-8, less significant P-value can be used. Alternatively, by providing pre-defined lead SNPs in a separate file, these provided SNPs will be defined as ind. sig. SNPs regardless of their P-value.
The higher the threshold for r2, the more SNPs are defined as ind. sig. SNPs. At the same time, the number of SNPs in the LD with the ind. sig. SNPs (the candidate SNPs; which are the SNPs annotated in FUMA and used for gene prioritization) decreases.
2. Lead SNPs
Lead SNPs are defined as SNPs which are ind. sig. SNPs and are independent from each other at r2 < 0.1 (from v1.3.5, this value can be specified by users). Therefore, lead SNPs are same as the SNPs clumped ind. sig. SNPs at the user defined P-value and r2 = 0.1 by plink.When r2 is set at 0.1, lead SNPs are exactly the same as ind. sig. SNPs. However, this will also result in selecting candidate SNPs that have r2 above 0.1 with any of ind. sig. SNPs. We thus advise to set r2 at 0.6 or higher.
3. Genomic risk loci
On top of lead SNPs, FUMA defines genomic risk loci, including all independent signals that are physically close or overlapping in a single locus. First, ind. sig. SNPs which are dependent each other at r2 ≥ 0.1 are assigned to the same genomic risk locus. Then, ind. sig. SNPs which are closer than the user defined distance (250 kb by default) are merged into one genomic risk locus. The distance between two LD blocks of two ind. sig. SNPs is the distance between the closest SNPs (which are in LD of the ind. sig. SNPs at user defined r2) from each LD block.Each locus is represented by the top lead SNP which has the minimum P-value in the locus.
4. Candidate SNPs (SNPs in LD of ind. sig. SNPs)
Candidate SNPs are SNPs that are in LD with any of the ind. sig. SNPs at the user defined r2. Candidate SNPs, together with the ind. sig. SNPs, are the SNPs that are used to prioritize genes. The most left and most right SNPs which are in LD of a ind. sig. SNP define a LD block in which those SNPs are used to compute distance between LD blocks.Note that not all SNPs are necessary in LD with lead SNPs, although they must be in LD with ind. sig. SNPs at the user defined r2.
All candidate SNPs are annotated and their functions and listed in the "SNPs" table.
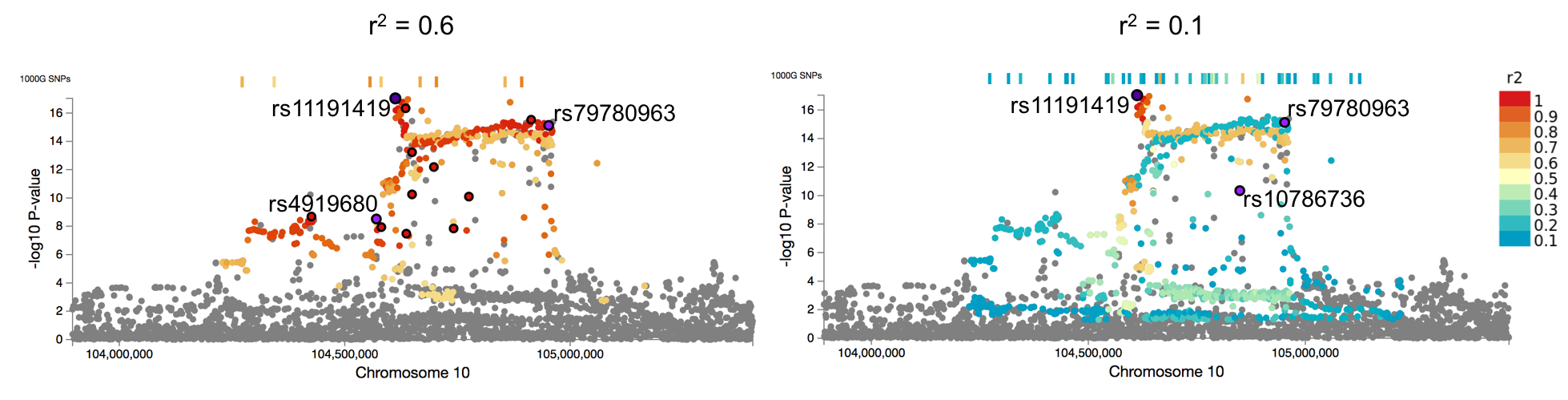
The higher the threshold r2, the less candidate SNPs are identified. The number of candidate SNPs can also be controlled by the parameter of the maximum P-value for gwas-tagged SNPs (0.05 by default). For example, when r2 is set at less than 0.6, a parameter of P-value threshold for GWAS tagged SNPs might need to be set at more significant since SNPs with r2 often have very high P-value.
Effect of r2 parameter
eQTLs
FUMA contains several data sources of eQTLs and each data source is described in this section.eQTL data sources
1. GTEx v6
Data source
eQTL data was downloaded from http://www.gtexportal.org/home/datasets.
Under the section of GTEx V6, from single tissue eQTL data both GTEx_analysis_V6_eQTLs.tar.gz
for significant SNP-gene association based on permutation, and
GTEx_Analysis_V6_all-snp-gene-associations.tar for every SNP-gene association test (including non-significant paris)
were downloaded.
GTEx eQTL v6 contains 44 different tissue types across 30 general tissue types.
Description
FUMA contains all SNP-gene pairs of cis-eQTL with nominal P-value < 0.05 (including non-significant associations).
Significant eQTLs are defined as FDR (gene q-value) ≤ 0.05.
The gene FDR is pre-calculated by GTEx and every gene-tissue pair has a defined P-value threshold for eQTLs based on permutation.
Signed statistics are t-statistics.
Samples
| General tissue type | Tissue type | Genotyped sample size |
|---|---|---|
| Adipose Tissue | Adipose Subcutaneous | 298 |
| Adipose Tissue | Adipose Visceral Omentum | 185 |
| Adrenal Gland | Adrenal Gland | 126 |
| Blood | Cells EBV-transformed lymphocytes | 114 |
| Blood Vessel | Artery Aorta | 197 |
| Blood Vessel | Artery Coronary | 118 |
| Blood Vessel | Artery Tibial | 285 |
| Blood | Whole Blood | 338 |
| Brain | Brain Anterior cingulate cortex BA24 | 72 |
| Brain | Brain Caudate basal ganglia | 100 |
| Brain | Brain Cerebellar Hemisphere | 89 |
| Brain | Brain Cerebellum | 103 |
| Brain | Brain Cortex | 96 |
| Brain | Brain Frontal Cortex BA9 | 92 |
| Brain | Brain Hippocampus | 81 |
| Brain | Brain Hypothalamus | 81 |
| Brain | Brain Nucleus accumbens basal ganglia | 93 |
| Brain | Brain Putamen basal ganglia | 82 |
| Breast | Breast Mammary Tissue | 183 |
| Colon | Colon Sigmoid | 124 |
| Colon | Colon Transverse | 169 |
| Esophagus | Esophagus Gastroesophageal Junction | 127 |
| Esophagus | Esophagus Mucosa | 241 |
| Esophagus | Esophagus Muscularis | 218 |
| Heart | Heart Atrial Appendage | 159 |
| Heart | Heart Left Ventricle | 190 |
| Liver | Liver | 97 |
| Lung | Lung | 278 |
| Muscle | Muscle Skeletal | 361 |
| Nerve | Nerve Tibial | 256 |
| Ovary | Ovary | 85 |
| Pancreas | Pancreas | 149 |
| Pituitary | Pituitary | 87 |
| Prostate | Prostate | 87 |
| Skin | Cells Transformed fibroblasts | 272 |
| Skin | Skin Not Sun Exposed Suprapubic | 196 |
| Skin | Skin Sun Exposed Lower leg | 302 |
| Small Intestine | Small Intestine Terminal Ileum | 77 |
| Spleen | Spleen | 89 |
| Stomach | Stomach | 170 |
| Testis | Testis | 157 |
| Thyroid | Thyroid | 278 |
| Uterus | Uterus | 70 |
| Vagina | Vagina | 79 |
2. Blood eQTL browser (Westra et al. 2013)
Data source
eQTL data was downloaded from http://genenetwork.nl/bloodeqtlbrowser/.
Description
The data only include eQTLs with FDR ≤ 0.5.
Genes in the original files were mapped to Ensembl ID in which genes are removed if they are not mapped to Ensembl ID.
Signed statistics are Z-scores.
Samples
5,311 peripheral blood samples from 7 studies (Westra et al. 2013).
3. BIOS QTL browser (Zhernakova et al. 2017)
Data source
eQTL data was downloaded from http://genenetwork.nl/biosqtlbrowser/.
Cis-eQTLs Gene-level all primary effects was downloaded which includes all SNP-gene pairs with FDR ≤ 0.05.
Description
The data only include eQTLs with FDR ≤ 0.05.
Signed statistics are betas.
Samples
2,116 whole peripheral blood samples of healthy adults from 4 Dutch cohorts (Zhernakova et al. 2017).
4. BRAINEAC
Data source
eQTL was obtained from http://www.braineac.org/.
Description
The data include all eQTLs with nominal P-value < 0.05.
Since tested allele was not provided in the original data source, minor alleles in 1000 genome phase 3 are assigned as tested alleles.
Signed statistics are t-statistics.
eQTLs were identified for each of the following 10 brain regions and based on averaged expression across all of them.
Alignment of risk increasing allele and eQTL tested allele was not performed for this data source,
since tested allele is not available in the original data source
(replaced with "NA" in the result table).
- Cerebellar cortex
- Frontal cortex
- Hippocampus
- Inferior olivary nucleus (sub-dissected from the medulla)
- Occipital cortex
- Putamen (at the level of the anterior commissure)
- Substantia nigra
- Temporal cortex
- Thalamus (at the level of the lateral geniculate nucleus)
- Intralobular white matter
Samples
134 neuropathologically confirmed control individuals of European descent from UK Brain Expression Consortium
(Ramasamy et al. 2014).
5. GTEx v7
Data source
eQTL data was downloaded from http://www.gtexportal.org/home/datasets.
Under the section of GTEx V7, from single tissue eQTL data both GTEx_analysis_v7_eQTLs.tar.gz
for significant SNP-gene association based on permutation, and
GTEx_Analysis_v7_all_associations.tar.gz for every SNP-gene association test (including non-significant pairs)
were downloaded.
GTEx eQTL v7 contains 53 different tissue types across 30 general tissue types.
Description
FUMA contains all SNP-gene pairs of cis-eQTL with nominal P-value < 0.05 (including non-significant associations).
Significant eQTLs are defined as FDR (gene q-value) ≤ 0.05.
The gene FDR is pre-calculated by GTEx and every gene-tissue pair has a defined P-value threshold for eQTLs based on permutation.
Signed statistics are betas.
Samples
| General tissue type | Tissue type | Genotyped sample size |
|---|---|---|
| Adipose Tissue | Adipose Subcutaneous | 385 |
| Adipose Tissue | Adipose Visceral Omentum | 313 |
| Adrenal Gland | Adrenal Gland | 175 |
| Blood | Cells EBV-transformed lymphocytes | 117 |
| Blood | Whole Blood | 369 |
| Blood Vessel | Artery Aorta | 267 |
| Blood Vessel | Artery Coronary | 152 |
| Blood Vessel | Artery Tibial | 388 |
| Brain | Brain Amygdala | 88 |
| Brain | Brain Anterior cingulate cortex BA24 | 109 |
| Brain | Brain Caudate basal ganglia | 144 |
| Brain | Brain Cerebellar Hemisphere | 125 |
| Brain | Brain Cerebellum | 154 |
| Brain | Brain Cortex | 136 |
| Brain | Brain Frontal Cortex BA9 | 118 |
| Brain | Brain Hippocampus | 111 |
| Brain | Brain Hypothalamus | 108 |
| Brain | Brain Nucleus accumbens basal ganglia | 130 |
| Brain | Brain Putamen basal ganglia | 111 |
| Brain | Brain Spinal cord cervical c-1 | 83 |
| Brain | Brain Substantia nigra | 80 |
| Breast | Breast Mammary Tissue | 251 |
| Colon | Colon Sigmoid | 203 |
| Colon | Colon Transverse | 246 |
| Esophagus | Esophagus Gastroesophageal Junction | 213 |
| Esophagus | Esophagus Mucosa | 358 |
| Esophagus | Esophagus Muscularis | 335 |
| Heart | Heart Atrial Appendage | 264 |
| Heart | Heart Left Ventricle | 272 |
| Liver | Liver | 153 |
| Lung | Lung | 383 |
| Muscle | Muscle Skeletal | 491 |
| Nerve | Nerve Tibial | 361 |
| Ovary | Ovary | 122 |
| Pancreas | Pancreas | 220 |
| Pituitary | Pituitary | 157 |
| Prostate | Prostate | 132 |
| Salivary Gland | Minor Salivary Gland | 85 |
| Skin | Cells Transformed fibroblasts | 300 |
| Skin | Skin Not Sun Exposed Suprapubic | 335 |
| Skin | Skin Sun Exposed Lower leg | 414 |
| Small Intestine | Small Intestine Terminal Ileum | 122 |
| Spleen | Spleen | 146 |
| Stomach | Stomach | 237 |
| Testis | Testis | 225 |
| Thyroid | Thyroid | 399 |
| Uterus | Uterus | 101 |
| Vagina | Vagina | 106 |
6. MuTHER (Grundberg et al. 2012)
Data source
eQTL data was downloaded from http://www.muther.ac.uk/.
Description
Chromosome coordinate was lifted over to hg19 from hg18 using liftOver software.
Gene names are mapped to Ensembl ID (excluded genes which are not mapped to ENSG ID).
Since only tested allele was provided, other allele was extracted from 1000G EUR population.
FDR (or any corrected P-value) was not available in the original data (in the FUMA, FDR column was replaced with NA).
Signed statistics are betas.
Since FDR is not available, MuTHER eQTLs can be only used when P-value threshold provided by user,
not "only significant snp-gene pairs" option.
Samples
856 female individuals of European descent recruited from
the TwinsUK Adult twin registry (Grundberg et al. 2012).
- Adipose (N=855)
- Skin (N=847)
- LCL (N=837)
7. xQTLServer (Ng et al. 2017)
Data source
eQTL data was downloaded from http://mostafavilab.stat.ubc.ca/xqtl/.
Description
Gene names are mapped to Ensembl ID (excluded genes which are not mapped to ENSG ID).
Since alleles were not available in the original data, extracted from 1000G EUR population based on chromosome coordinate.
FDR was not provided in the original data source, but the FDR column was replaced with Bonferroni corrected p-value,
as it was used in the original study (corrected for all tested SNP-gene pairs 60,456,556).
Signed statistics are not available.
Alignment of risk increasing allele and eQTL tested allele was not performed for this data source,
since tested allele and signed statistics are not available in the original data source
(replaced with "NA" in the result table).
Samples
494 dorsolateral prefrontal cortex samples (Ng et al. 2017).
8. CommonMind Consortium (Fromer et al. 2016)
Data source
eQTL data was downloaded from https://www.synapse.org//#!Synapse:syn5585484.
Both eQTLs with and without SVA are included.
Description
Publicly available eQTLs from CMC (without application) is binned by FDR.
Therefore, nominal P-value is not available (replaced with NA).
FDR was binned into the following four groups, <0.2, <0.1, <0.05 and <0.01.
As numeric value is required for filtering during SNP2GENE process, those categorical values are replaced with
0.199, 0.099, 0.049 and 0.009 respectively.
Signed statistics are not available but since expressed increasing allele was provided, signed_stats column is replaced with 1.
Trans eQTLs are also available for CMC data set (as a separated option from cis-eQTLs).
Samples
Post-mortem brain samples from 467 Caucasian individuals (209 with SCZ, 206 controls and 52 AFF cases; Fromer et al. 2016).
9. eQTLGen (Vosa et al. 2018)
Data source
eQTL data was downloaded from http://www.eqtlgen.org/index.html.
For cis-eQTLs, cis-eQTLs_full_20180905.txt.gz,
for trans-eQTLs, trans-eQTL_significant_20181017.txt.gz was used.
Description
Full summary statistics were downloaded.
For cis-eQTLs, full summary statistics was downloaded.
In the dataset, every SNP-gene pair with a distance <1Mb from the center of the gene and tested in at least 2 cohorts was included.
For trans-eQTLs, only significant eQTLs were included in FUMA since the cross-mapping effects were not filtered in the downloadable full summary statistics.
In the original study, every SNP-gene pair with a distance >5Mb and tested in at least 2 cohorts was included.
FDR was estimated based on permutations.
Please refer the original study for more details (Vosa et al. 2018).
Ensembl gene ID is used as provided in the original file.
Signed statistics are z-scores.
Samples
Meta-analysis of cis-/trans-eQTLs from 37 datasets with a total of 31,684 individuals.
10. PsychENCODE (Wang et al. 2018)
Data source
eQTL data was downloaded from http://resource.psychencode.org.
We used significant (DER-08a_hg19_eQTL.significant).
Description
The available eQTLs were filtered based on an FDR <0.05 and an expression >0.1 FPKM in at least 10 samples.
Please refer the original study for more details (Wang et al. 2018).
Ensembl gene ID is used as provided in the original file.
The signed statistics are betas.
Samples
The eQTLs were identified from 1387 individuals.
11. DICE (Schmiedel et al. 2018)
Data source
eQTL data was downloaded from https://dice-database.org/downloads#eqtl_download.
The cis-eQTLs were obtained from the DICE eQTL section of the website.
Description
Only significant eQTLs are present in the dataset.
The available eQTLs were filtered based on FDR<0.05, nominal P-value<0.0001, and TPM>0.1.
FDR was estimated using permutation.
Please refer the original study for more details (Schmiedel et al. 2018).
Ensembl gene ID is used as provided in the original file.
FDR was not provided in the original source, but since the eQTLs were already filtered on FDR<0.05
all eQTLs were assigned to FDR 0.049 to be able to pass the filtering of the "only significant snp-gene pairs" option.
Signed statistics are betas.
The cell types were:
- Naive B cells
- Activated CD4 T cells
- Naive CD4 T cells
- Activated CD8 T cells
- Naive CD8 T cells
- Classical Monocytes
- Non-classical Monocytes
- NK cell, CD56dim CD16+
- TFH CD4 T cells
- TH117 CD4 T cells
- TH1 CD4 T cells
- TH2 CD4 T cells
- Memory TREG CD4 T cells
- Naive TREG CD4 T cells
Samples
The eQTLs were identified in 13 immune cell types isolated from 106 leukapheresis samples provided by 91 healthy subjects.
12. van der Wijst et al. scRNA eQTLs (van der Wijst et al. 2018)
Data source
eQTL data was downloaded from https://molgenis26.target.rug.nl/downloads/scrna-seq/.
Description
The tested allele was specified in the data, but the other allele was not.
FDR was estimated using permutation.
Please refer the original study for more details (van der Wijst et al. 2018).
Ensembl gene ID is used as provided in the original file.
The summary statistics are Z scores.
The cell types were:
- B cells
- CD4 T cells
- CD8 T cells
- Peripheral blood mononuclear cells (PBMC)
- Monocytes
- Classical monocytes
- Non-classical monocytes
- Natural killer (NK) cells
- Dendritic cells (DC)
Samples
The eQTLs were identified from 25,000 peripheral blood mononuclear cells (PBMCs) from 45 donors.
13. GTEx v8
Data source
eQTL data was downloaded from http://www.gtexportal.org/home/datasets.
Under the section of GTEx V8, from single tissue eQTL data both GTEx_Analysis_v8_eQTL.tar
for significant SNP-gene associations, and all tested pairs of SNP-gene were obtained from GCP (including non-significant pairs).
GTEx eQTL v8 contains 54 different tissue types across 30 general tissue types.
Description
FUMA contains all SNP-gene pairs of cis-eQTL with nominal P-value < 0.05 (including non-significant associations).
Significant eQTLs are defined as FDR (gene q-value) ≤ 0.05.
The gene FDR is pre-calculated by GTEx and every gene-tissue pair has a defined P-value threshold for eQTLs based on permutation.
Signed statistics are betas.
Samples
| General tissue type | Tissue type | Genotyped sample size |
|---|---|---|
| Adipose Tissue | Adipose Subcutaneous | 581 |
| Adipose Tissue | Adipose Visceral Omentum | 469 |
| Adrenal Gland | Adrenal Gland | 233 |
| Blood | Cells EBV-transformed lymphocytes | 147 |
| Blood | Whole Blood | 670 |
| Blood Vessel | Artery Aorta | 387 |
| Blood Vessel | Artery Coronary | 213 |
| Blood Vessel | Artery Tibial | 584 |
| Brain | Brain Amygdala | 129 |
| Brain | Brain Anterior cingulate cortex BA24 | 147 |
| Brain | Brain Caudate basal ganglia | 194 |
| Brain | Brain Cerebellar Hemisphere | 175 |
| Brain | Brain Cerebellum | 209 |
| Brain | Brain Cortex | 205 |
| Brain | Brain Frontal Cortex BA9 | 175 |
| Brain | Brain Hippocampus | 165 |
| Brain | Brain Hypothalamus | 170 |
| Brain | Brain Nucleus accumbens basal ganglia | 202 |
| Brain | Brain Putamen basal ganglia | 170 |
| Brain | Brain Spinal cord cervical c-1 | 126 |
| Brain | Brain Substantia nigra | 114 |
| Breast | Breast Mammary Tissue | 396 |
| Colon | Colon Sigmoid | 318 |
| Colon | Colon Transverse | 368 |
| Esophagus | Esophagus Gastroesophageal Junction | 330 |
| Esophagus | Esophagus Mucosa | 497 |
| Esophagus | Esophagus Muscularis | 465 |
| Heart | Heart Atrial Appendage | 372 |
| Heart | Heart Left Ventricle | 386 |
| Kidney | Kidney Cortex | 73 |
| Liver | Liver | 208 |
| Lung | Lung | 515 |
| Muscle | Muscle Skeletal | 706 |
| Nerve | Nerve Tibial | 532 |
| Ovary | Ovary | 167 |
| Pancreas | Pancreas | 305 |
| Pituitary | Pituitary | 237 |
| Prostate | Prostate | 221 |
| Salivary Gland | Minor Salivary Gland | 144 |
| Skin | Cells Clustured fibroblasts | 483 |
| Skin | Skin Not Sun Exposed Suprapubic | 517 |
| Skin | Skin Sun Exposed Lower leg | 605 |
| Small Intestine | Small Intestine Terminal Ileum | 174 |
| Spleen | Spleen | 227 |
| Stomach | Stomach | 324 |
| Testis | Testis | 322 |
| Thyroid | Thyroid | 574 |
| Uterus | Uterus | 142 |
| Vagina | Vagina | 156 |
14. eQTL Catalogue
Data source
eQTL data was downloaded from the eQTLcatalogue (not from the original data source).
The paths to individual datasets can be found at https://github.com/eQTL-Catalogue/eQTL-Catalogue-resources/blob/master/tabix/tabix_ftp_paths.tsv.
Only the gene level (ge) files were included.
Details of each dataset are described below.
Datasets which were already present on FUMA have not been included (DICE & xQTLServer).
As of FUMA v1.6.1, only nominally significant (P<0.05) eQTLs identified in the data were included.
Description
The eQTLs were mapped to hg19 from hg38 using liftOver software.
Significant eQTLs are defined using a nominal p-value (0.00001).
More information on the methods used to generate the eQTL data can be found at https://www.ebi.ac.uk/eqtl/Methods/.
Datasets
| Dataset | Pubmed ID | Tissue types | Conditions | Sample size (Samples/Donors) |
|---|---|---|---|---|
| Alasoo_2018 | 29379200 | Macrophage | Naive, IFNg, Salmonella, IFNg + Salmonella | 336/84 |
| BLUEPRINT | 27863251 | Monocytes, neutrophils, T-cells | 554/197 | |
| BrainSeq | 30050107 | Dorsolateral prefrontal cortex | 484/484 | |
| CEDAR | 29930244 | CD4 and CD8 T-cells, monocytes, neutrophils, platelet, B-cells, ileum, rectum, transverse colon | 2338/322 | |
| Fairfax_2012 | 22446964 | B-cells | 282/282 | |
| Fairfax_2014 | 24604202 | Monocytes | Naive, IFN24, LPS2, LPS24 | 1372/424 |
| GENCORD | 23755361 | Lymphoblastoid cell lines, fibroblasts, T-cells | 560/195 | |
| GEUVADIS | 24037378 | Lymphoblastoid cell lines | 445/445 | |
| HipSci | 28489815 | iPSCs | 322/322 | |
| Kasela_2017 | 28248954 | CD4 and CD8 T-cells | 533/297 | |
| Lepik_2017 | 28922377 | Blood | 491/491 | |
| Naranbhai_2015 | 26151758 | Neutrophils | 93/93 | |
| Nedelec_2016 | 27768889 | Macrophages | Naive, Listeria, Salmonella | 493/168 |
| Quach_2016 | 27768888 | Monocytes | Naive, LPS, Pam3CSK4, R848, IAV | 969/200 |
| Schwartzentruber_2018 | 29229984 | Sensory neurons | 98/98 | |
| TwinsUK | 25436857 | Fat, Lymphoblastoid cell lines, skin, blood | 1364/433 | |
| van_de_Bunt_2015 | 26624892 | Pancreatic islets | 117/117 |
15. EyeGEx
Data source
eQTL data was downloaded from the GTEx website https://gtexportal.org/home/datasets. The file containing the cis-eQTLs can be downloaded from https://storage.googleapis.com/gtex_external_datasets/eyegex_data/single_tissue_eqtl_data/Retina.nominal.eQTLs.with_thresholds.tar.
All eQTLs identified in the data were included.
Description
Please refer to the original study for more details (Ratnapriya et al. 2019).
Ensembl gene ID is used as provided in the original file. FDR adjusted P-values were calculated based on gene-level FDR threshold.
The signed statistics are betas.
Samples
The eQTLs were identified from 406 individuals.
16. InsPIRE
Data source
eQTL data was downloaded from zenodo https://zenodo.org/record/3408356. The file containing the cis-eQTLs can be downloaded from https://zenodo.org/record/3408356/files/InsPIRE_Islets_Gene_eQTLs_Nominal_Pvalues.txt.gz?download=1.
All nominally significant (P<0.05) eQTLs identified in the data were included except two variant-gene connections which were reported twice (5:150176501:C:A-ENSG00000197083.7 and 5:150176501:C:A-ENSG00000211445.7).
Description
Human pancreatic islets were the tested tissue.
Please refer to the original study for more details (Viñuela et al. 2020).
Ensembl gene ID is used as provided in the original file after the version number was removed (e.g. ENSG00000211445.7 became ENSG00000211445). The FDR reported in the paper could not be calculated using the downloaded data. Instead the FDR value was set to 1 for all variants except those found to be independently significant (included in PacreaticIslets_independent_gene_eQTLs.txt). The FDR value of the independently significant variants was set to 1e-5.
The signed statistics are betas.
Samples
The eQTLs were identified from 420 individuals.
17. TIGER
Data source
eQTL data was downloaded from the TIGER website http://tiger.bsc.es/downloads. The file containing the cis-eQTLs can be downloaded from http://tiger.bsc.es/assets/tiger_eqtl_stats.tar.gz.
All nominally significant (P<0.05) eQTLs identified in the data were included. The eQTLs on the X chromosome were not included because the X chromosome was analysed separately for males and females, which would have led to duplicate measurements for the same variant-gene connection.
Description
Human pancreatic islets were the tested tissue.
Please refer to the original study for more details (Alonso et al. 2021).
Ensembl gene ID is used as provided in the original file. FDR values were calculated by adjusting the P-values so that an FDR of 0.05 was equal to a P-value of 6.2e-4. This was the significance threshold described in the original paper.
The signed statistics are Z-scores.
Samples
The eQTLs were identified from 404 individuals.
Alignment of risk increasing allele in GWAS and tested allele of eQTLs
Risk increasing allele in GWAS
When "beta" or "OR" column is provided in the input GWAS file, risk increasing alleles are defined as follows:
if beta > 0 or OR > 1, effect/risk allele is defined as the risk increasing allele,
if beta < 0 or OR < 1, non-effect/non-risk allele is defined as the risk increasing allele.
If signed effect is not provided in the input GWAS file, risk increasing allele is not defined ("NA").
SNPs which are not in the input GWAS file but obtained from reference panel due to high LD are also encoded as "NA".
When both effect and non-effect alleles are not provided in the input GWAS file, this alignment is not relevant.
Please be careful to interpret the results.
Aligned direction of eQTLs
The sign of the t-statistics or z-score of the original eQTL data sources represents the direction of effect of tested allele.
To obtain the direction of effect for risk increasing allele of GWAS, risk increasing allele and tested allele of eQTLs are aligned as follows:
if risk increasing allele is the same allele as tested allele of the eQTL, direction is the same as the sign of the original t-statistics/z-score,
if risk increasing allele is not same allele as tested allele of the eQTL, direction of t-statistics/z-score was flipped.
Direction is either "+" (risk increasing allele increases the expression of the gene) or "-" (risk increasing allele decreases the expression of the gene).
Examples
Here are some examples how the alleles are aligned.
| uniqID | effect allale | non-effect allele | beta | risk increasing allele | tested allele of eQTL | t-statistics of eQTL | aligned direction |
|---|---|---|---|---|---|---|---|
| 1:201885026:C:T | T | C | 0.22 | T | T | -7.98 | - |
| 11:43843579:C:G | C | G | 0.004 | C | G | 17.23 | - |
| 16:28537971:C:T | T | C | -0.028 | C | C | 5.04 | + |
Chromatin interaction data and mapping
In this section, build in chromatin interaction data, file format of custom chromatin interaction matrices and details of chromatin interaction mapping are described. Since chromatin interaction mapping is more complicated than other two mappings (positional and eQTL), please read this section carefully.Terminology
Region 1
One end of a significant interaction which overlap with one of the candidate SNPs (independent significant SNPs and SNPs which are in LD of them).
This region is always overlap with one of the genomic risk loci identified by FUMA.
Region 2
Another end of the significant interaction.
This region is used to map to genes.
Region 2 could also be overlapped with one of the genomic risk loci.
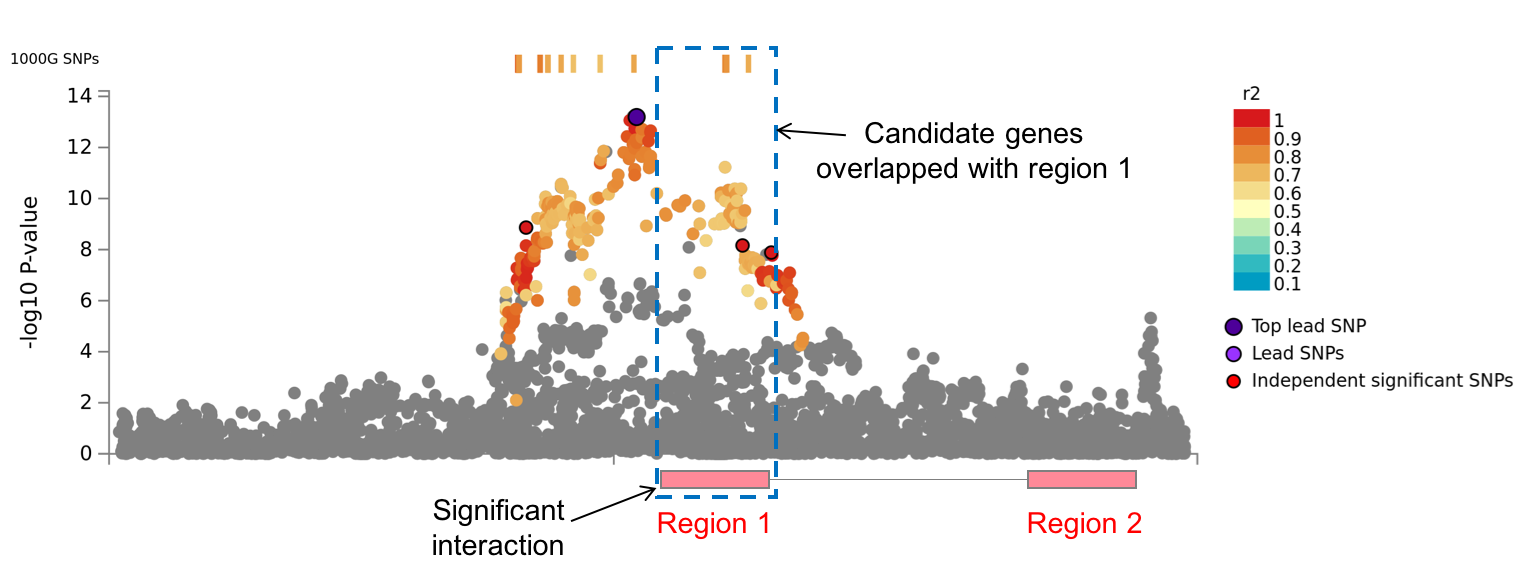
Direction of interactions
Input files of chromatin interaction consist of 7 columns: chr1, start1, end1, chr2, start2, end2, FDR (or other score). For loops identified by HiC, there is no directionality, i.e. both directions (chr1:start1-end1 <-> chr2:start2-end2) were considered regardless of the order in the file. For enhancer-promoter (EP) links, only one way (enhancer -> promoter) is considered for the mapping. The directionality is specified for each dataset below.
Build in chromatin interaction data
1. Hi-C of 21 tissue/cell types from GSE87112.
Pre-processed significant loops computed by Fit-Hi-C were obtained from GSE87112.
Loops were filtered at FDR 0.05. For mapping, loops can be further filter by the user defined FDR threshold.
Both directions are considered.
Available tissue/cell types are listed below.
- Adrenal
- Aorta
- Bladder
- Dorsolateral Prefrontal Cortex
- Hippocampus
- Left Ventricle
- Liver
- Lung
- Ovary
- Pancreas
- Psoas
- Right Ventricle
- Small Bowel
- Spleen
- GM12878
- IMR90
- Mesenchymal Stem Cell
- Mesendoderm
- Neural Progenitor Cell
- Trophoblast-like Cell
- hESC
2. Hi-C loops from Giusti-Rodriguez et al. 2019
Pre-processed enhancer-promoter and promoter-promoter interactions based on
HiC data for adult and fetal human brain samples.
The data was provided by Prof. Patric F. Sullivan.
Only significant interaction with P < 2.31e-11 (after Bonferroni correction) were included.
Both directions are considered.
3. Hi-C based data from PsychENCODE
3.1 Enhancer-Promoter links based on Hi-C
The data was downloaded from PsychENCODE resource
(file: INT-16_HiC_EP_linkages.csv).
Promoter regions were defined as 1000 around the provided TSS site.
Since there is no P-value/FDR/score, all interactions were assigned to 0.
Only one way (enhancer -> promoter) is considered.
3.2 Promoter anchored Hi-C loops
The data was downloaded from PsychENCODE resource
(file: Promoter-anchored_chromatin_loops.bed).
Since there is no P-value/FDR/score, all interactions were assigned to 0.
Only one way (region -> promoter) is considered.
4. Enhancer-Promoter correlations from FANTOM5 The data was downloaded from FANTOM5 human Enhancer Tracks (file: hg19_enhancer_promoter_correlations_distances_cell_type.txt and hg19_enhancer_promoter_correlations_distances_organ.txt). Only one way (enhancer -> promoter) is considered.
Custom chromatin interaction matrices file format
1. Input file format
The chromatin interaction files should have the following 7 columns in the order as listed below.
Header line is mandatory but the column names do not need to be the same as the below as long as the order is the same.
Delimiter should be tab or white space(s).
The input file should be gzipped and named as "(name_of_data).txt.gz" in which "(name_of_data)" will be used in the result table and regional plot.
Columns:
- chromosome of region 1
- start position of region 1
- end position of region 1
- chromosome of region 2
- start position of region 2
- end position of region 2
- FDR
chr1 start1 end1 chr2 start2 end2 FDR
1 2920001 2960000 1 3160001 3200000 0.03186403
1 4160001 4200000 1 5880001 5920000 5.3e-8
1 4520001 4560000 3 83200001 83240000 0.03920674
Chromosome can be coded as string like "chr1" and "chrX" which will be converted into integer.
Order of regions does not matter, unless a word "oneway" is in the file name (e.g. hic_loops_oneway.txt.gz). In that case only one direction (1st region -> 2nd region) is considered.
Inter-chromosomal interactions can be encoded in the same file by specifying chromosome of region 1 and region 2.
The column of FDR will be used to filter interaction by the user defined threshold.
The maximum size of each file is 600Mb. If the file is larger than this, please filter interactions or split them into multiple files.
2. Data types
When uploading custom chromatin interaction matrices, users can specify the type of data such as Hi-C or ChIA-PET. Specifying the data type is not mandatory since it is only used to specify in the result table and regional plot for convenience.
3. Filtering of chromatin interactions
The 7th column (FDR) will be used to filter interactions. To prevent from this filtering, either set filtering threshold to 1 or assign 0 to the FDR column. Technically, the 7th column does not have to be FDR but any other scores. When one prefers to use different score or nominal P-value, that is also possible by setting proper filtering threshold. Note that, interactions will be filtered on which have score less than or equal to the threshold.
Enhancer and promoter regions
Enhancer and promoter regions were obtained from Roadmap Epigenomics Projects for 111 epigenomes.
Those regions were predicted using DNase peaks and core 15-state chromatin state model.
Please refer here for details.
For selected epigenomes, enhancer regions are annotated to region 1 and promoter regions are annotated to region 2.
Dyadic enhancer/promoter regions are annotated for both.
Annotated enhancer and promoter regions can be used to filter SNPs or mapped genes which is described in the next section.
Chromatin interaction mapping
1. Basic mapping (without filtering)
Chromatin interaction mapping is performed with significant chromatin interactions at the user defined threshold.
Regions 2 is mapped to genes whose promoter regions (250bp up- and 50bp down-stream of the TSS by default) are overlapped with the region 2.
Those genes were considered as mapped by candidate SNPs which are overlapped with region 1.
In the case there is not genes in region 2, those interactions are not mapped to any genes.
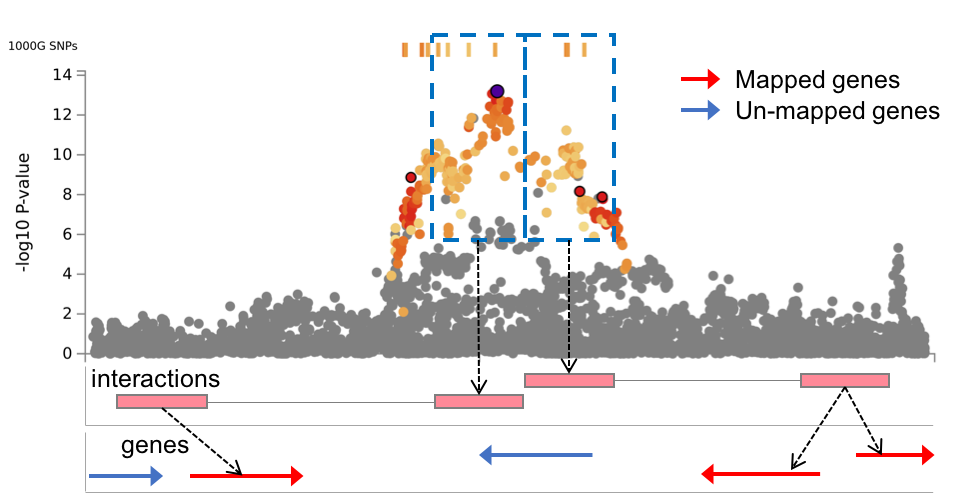
2. Enhancer filtering
When enhancers are annotated to region 1, user can select the option to filter candidate SNPs on such that are overlapped with enhancer regions of selected epigenomes.
Note that, in the result table, all significant interactions are included but not all are necessary used for mapping.
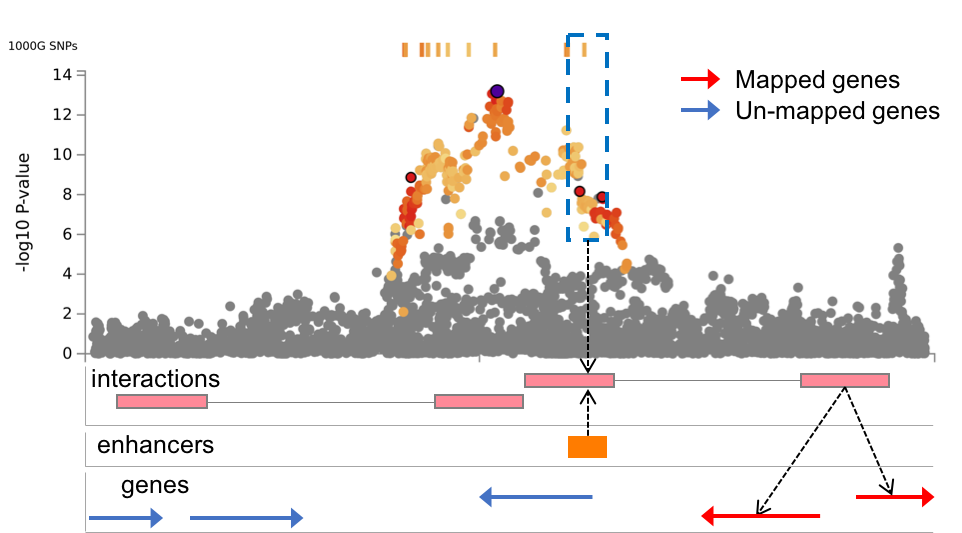
3. Promoter filtering
When promoters are annotated to region 2, user can select the option to limit the chromatin interaction mapping to only genes whose promoter regions are overlapped with annotated promoter regions of selected epigenomes.
Note that, in the result table, all significant interactions are included but not all are necessary mapped to genes.
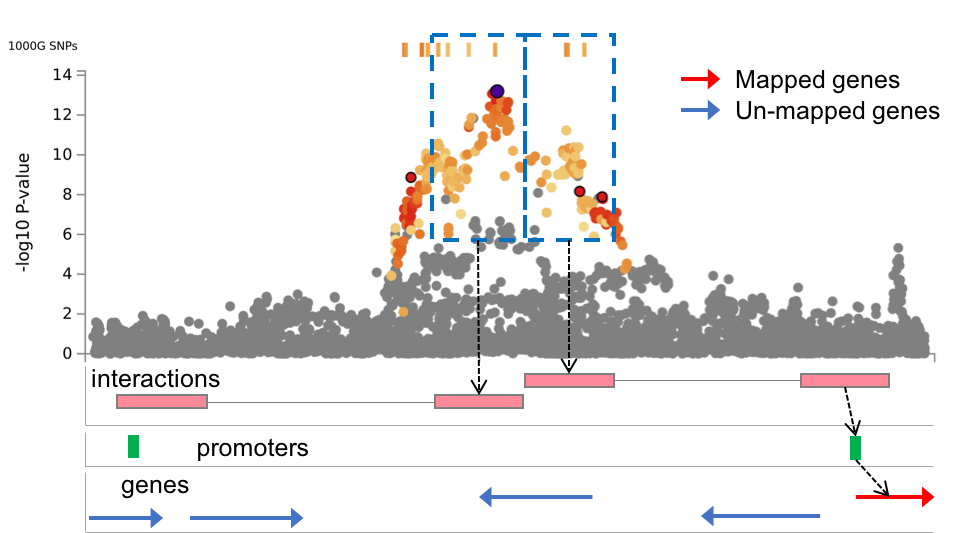
In very rare cares, when the promoter filtering is activated, genes whose promoter regions (250bp up- and 500bp down-stream of TSS) do not overlap with region 2 but do overlap with promoters from Roadmap that are overlapping with region 2 are mapped.
In this case, these genes are not in "ci.txt" file but in "ciProm.txt" file which can be linked to "ci.txt" by region 2.
GENE2FUNC
The main goal of GENE2FUNC is to provide information on expression of prioritized genes and test for enrichment of the set of genes in pre-defined pathways. You can use the genes prioritized with SNP2GENE or use a separate list of genes.
Submit genes
Option 1. Use mapped genes from SNP2GENE
If you want to use mapped genes from SNP2GENE, just click a button in Mapped genes panel of the result page. It will open a new tab and automatically starts analyses. This will take all mapped genes and use background genes with selected gene types for gene mapping (such as "protein-coding" or "ncRNA"). The method for multiple testing correction (FDR BH), adjusted P-value cutoff (0.05) and minimum number of overlapped genes (2) are set at default values. These options can be adjusted by resubmitting your query (click "Submit" button in New Query tab).
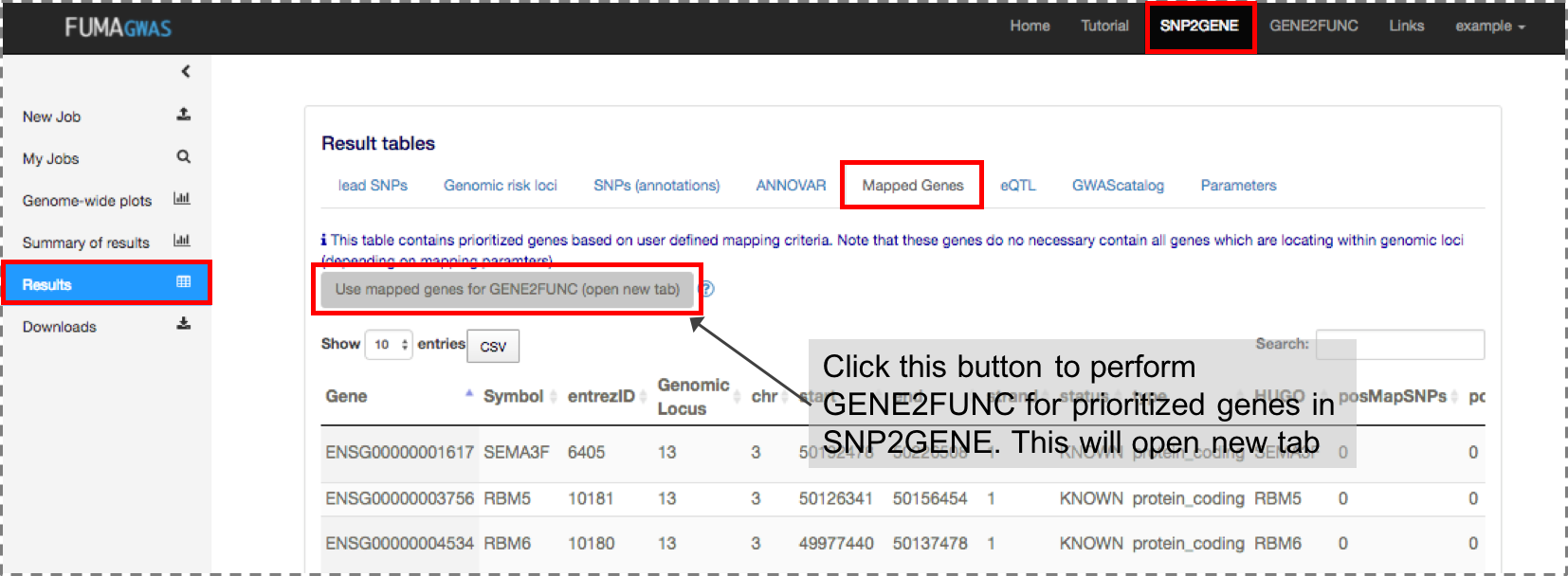
Option 2. Use a list of genes of interest
To analyze a custom list of genes, you have to prepare a list of genes as either ENSG ID, entrez ID or gene symbol. Genes can be provided in the text are (one gene per line) or by uploading a file in the left panel. When you upload a file, genes have to be in the first column with a header. Header can be anything (even just a new line is fine) but FUMA will start reading your genes from the second row.
To analyze your genes, you need to specify background genes, which are used in the 2x2 enrichment tests. You can choose from the provided gene types. Alternatively, you can provide a custom list of background genes. Please provide this list either in the text area or by uploading a file of the right panel. File format should be the same as described for genes of interest.
Outputs of GENE2FUNC
1. Summary of input genes and download files
1) Summary of input genes
The table summarised the input genes and background genes.
Input genes which are not used in the GENE2FUNC analyses due to lack of matching gene ID
are also listed.
Since the primary gene ID of FUMA is Ensembl ID and not all Ensembl IDs are mapped to unique
entrez ID (NCBI gene ID), the number of unique entrez ID can be smaller than the number of
input genes with Ensembl ID.
Ensembl ID is used for expression heatmap and tissue specificity analyses,
and entrez ID is used for gene set enrichment analysis.
2) Download files
Results of GENE2FUNC can be downloaded as text file from here.
3) Parameters
The table contains input parameters. This can be also downloaded from the option above.
2. Gene Expression Heatmap
The heatmap displays two expression values.
1) Average expression per label
This is an averaged expression value per label (e.g. tissue types or developmental stage)
per gene following to winsorization at 50 and log 2 transformation with pseudocount 1.
The expression value depends on the data set, RPKM (Read Per Kilobase per Million)
for GTEx v6 and BrainSapn, TPM (Transcripts Per Million) for GTEx v7.
This allows for comparison across labels and genes.
Hence, cells filled in red represent higher expression compared to cells filled in blue across genes and labels.
2) Average of normalized expression per label
This is the average of normalized expression (zero mean across samples)
following to winsorization at 50 and log 2 transformation of the expression value with pseudocount 1.
This allows comparison of gene expression across labels (horizontal comparison) within a gene.
Thus expression values of different genes within a label (vertical comparison) are not comparable.
Hence, cells filled in red represents higher expression of the genes in
a corresponding label compared to other labels, but it DOES NOT represent
higher expression compared to other genes.
Labels (columns) and genes (rows) can be ordered by alphabetically or cluster (hierarchical clustering).
Hierarchical clustering is performed using python scipy package (using "average" method).
The heatmap is downloadable in several file formats. Note that the image will be downloaded as displayed.
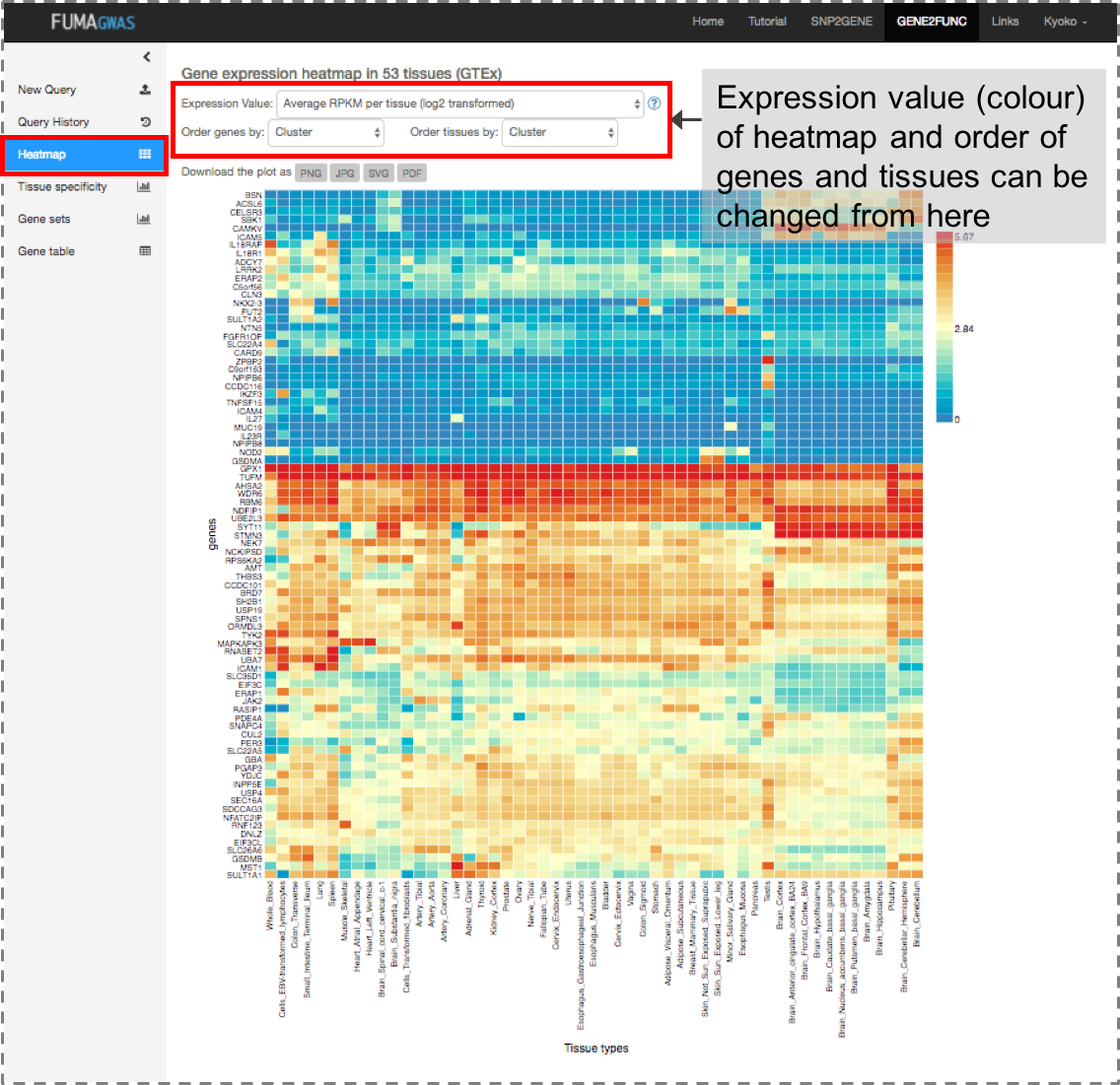
3. Tissue specificity
Tissue specificity is tested using the differentially expressed genes
defined for each label of each expression data set
Differentially Expressed Gene (DEG) Sets
DEG sets were pre-calculated by performing two-sided t-test for any one of labels against all others.
For this, expression values were normalized (zero-mean) following to a log 2 transformation of expression value (EPKM or TPM).
Genes which with P-value ≤ 0.05 after Bonferroni correction and absolute log fold change ≥ 0.58 were
defined as differentially expressed genes in a given label compared to others.
On top of DEG, up-regulated DEG and down-regulated DEG were also pre-calculated by taking sign of t-statistics into account.
Input genes were tested against each of the DEG sets using the hypergeometric test.
The background genes are genes that have average expression value > 1 in at
least one of the labels and exist in the user selected background genes.
Significant enrichment at Bonferroni corrected P-value ≤ 0.05 are coloured in red.
Note that for DEG sets, Bonferroni correction is performed for each of up-regulated, down-regulated and both-sided DEG sets separately.
Results and images are downloadable as text files and in several image file formats.
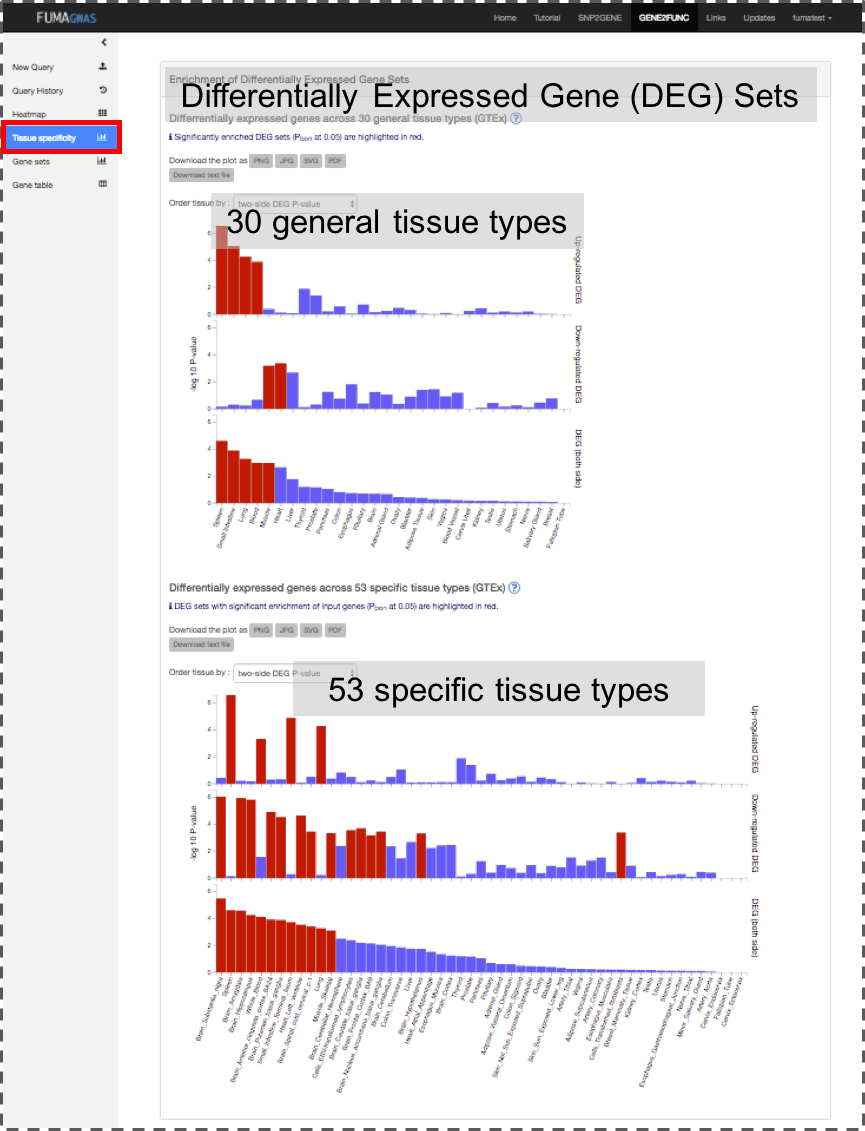
4. Gene Sets
Hypergeometric tests are performed to test if genes of interest are overrepresented in any of the pre-defined gene sets.
Multiple test correction is performed per category, (i.e. canonical pathways, GO biological processes and so on, separately).
Gene sets were obtained from MSigDB, WikiPathways and reported genes from the GWAS-catalog.
The MSigDB and WikiPathways data were downloaded with entrez IDs and included without modification.
The GWAS catalog data was downloaded with gene symbols and then converted to entrez ID using biomaRt.
If a single gene symbol matched multiple entrez IDs, then all matching entrez IDs were included in the geneset.
The following files were used to make the GENE2FUNC genesets:
| GENE2FUNC name | File used |
|---|---|
| Hallmark gene sets (MsigDB h) | h.all.v2023.1.Hs.entrez.gmt |
| Positional gene sets (MsigDB c1) | c1.all.v2023.1.Hs.entrez.gmt |
| Curated_gene_sets | c2.all.v2023.1.Hs.entrez.gmt |
| Chemical and Genetic pertubation gene sets (MsigDB c2) | c2.cgp.v2023.1.Hs.entrez.gmt |
| All Canonical Pathways (MsigDB c2) | c2.cp.v2023.1.Hs.entrez.gmt |
| BioCarta (MsigDB c2) | c2.cp.biocarta.v2023.1.Hs.entrez.gmt |
| KEGG (MsigDB c2) | c2.cp.kegg.v2023.1.Hs.entrez.gmt |
| Reactome (MsigDB c2) | c2.cp.reactome.v2023.1.Hs.entrez.gmt |
| microRNA targets (MsigDB c3) | c3.mir.v2023.1.Hs.entrez.gmt |
| TF targets (MsigDB c3) | c3.tft.v2023.1.Hs.entrez.gmt |
| All computational gene sets (MsigDB c4) | c4.all.v2023.1.Hs.entrez.gmt |
| Cancer gene neighborhoods (MsigDB c4) | c4.cgn.v2023.1.Hs.entrez.gmt |
| Cancer gene modules (MsigDB c4) | c4.cm.v2023.1.Hs.entrez.gmt |
| GO biological processes (MsigDB c5) | c5.go.bp.v2023.1.Hs.entrez.gmt |
| GO cellular components (MsigDB c5) | c5.go.cc.v2023.1.Hs.entrez.gmt |
| GO molecular functions (MsigDB c5) | c5.go.mf.v2023.1.Hs.entrez.gmt |
| Oncogenic signatures (MsigDB c6) | c6.all.v2023.1.Hs.entrez.gmt |
| Immunologic signatures (MsigDB c7) | c7.all.v2023.1.Hs.entrez.gmt |
| WikiPathways | c2.cp.wikipathways.v2023.1.Hs.entrez.gmt |
| Cell_type_signature (MSigDB c8) | c8.all.v2023.1.Hs.entrez.gmt |
The genesets used in the GENE2FUNC module can be downloaded here:
22M
29M
The full results are downloadable as a text file at the top of the page.
In each category, plot view and table view are selectable.
In the plot view, images are downloadable in several file formats.
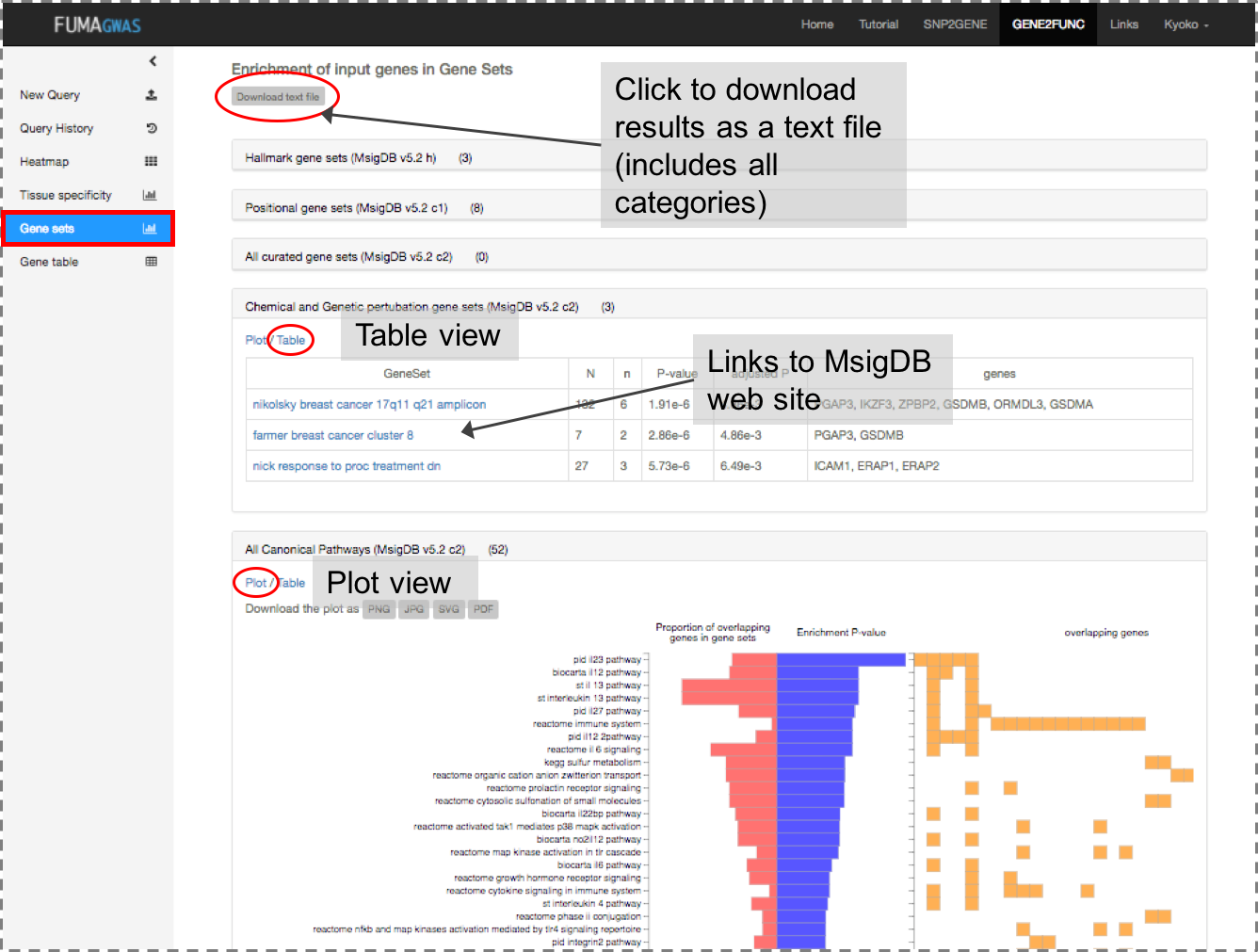
5. Gene Table
Input genes are mapped to OMIM ID, UniProt ID, Drug ID of DrugBank and links to GeneCards.
Drug IDs are assigned if the UniProt ID of the gene is one of the targets of the drug.
Each link to OMIM, Drugbank and GeneCards will open in a new tab.
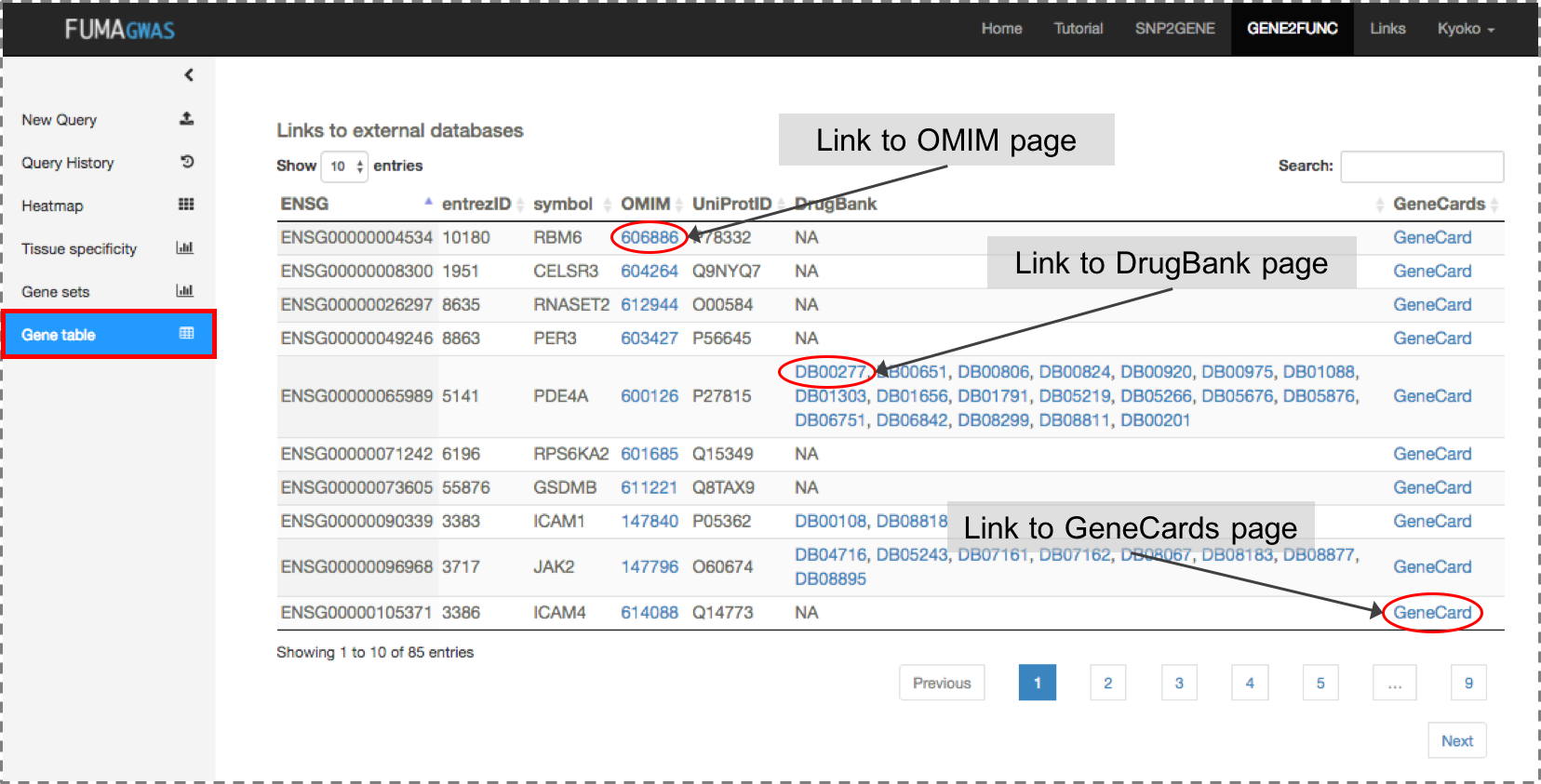
Cell type specificity analyses with scRNA-seq
How to perform cell type specificity analysis on FUMA
We use MAGMA gene-property analysis to test cell type specificity of phenotype with GWAS summary statistics. As an input, it requires XXX.genes.raw file from MAGMA gene analysis. You can either select your existing SNP2GENE job or upload MAGMA output file you run by yourself.FUMA uses Ensembl gene ID for all scRNA-seq data. If the input file contains different gene ID or gene symbols, FUMA will map to Ensembl gene ID. To do so, please UNCHECK the option right below the file selection, "Ensembl gene ID is used in the provided file". Otherwise MAGMA will result in an error due to mismatch of gene ID.
From FUMA v1.3.4, a 3-step workflow is implemented for the cell type analysis. This workflow consists of
- Step 1: per dataset analysis (same as implemented in v1.3.4)
After multiple testing correction across selected datasets, significant cell types were retained for Step 2. - Step 2: within dataset conditional analysis
Identify independent signals per dataset by performing forward-selection. - Step 3: cross datasets conditional analysis
Cell types retained from Step 2 are further conditioned each other across datasets to disentangle relationship between association of cell types from different datasets.
Note that step 2 and step 3 are not activated by default. To perform entire workflow, please CHECK the options.
MAGMA gene-property analysis with scRNA-seq
The gene-property analysis aims to test relationships between cell specific gene expression profiles and disease-gene associations. The gene-property analysis is based on the regression model, $$Z = \beta_0 + E_c\beta_E + A\beta_A + B\beta_B + \epsilon$$ where \(Z\) is a gene-based Z-score converted from the gene-based P-value, \(B\) is a matrix of several technical confounders included by default. \(E_c\) is the gene expression value of a testing cell type c and \(A\) is the average expression across cell types in a data set, defined as follows: $$E_c = \sum_{i}^{n} log_2(e_i + 1)/n$$ $$A = \sum_{j \in C}^{N} E_j/N$$ where \(n\) is the number of cells in the cell type c, \(e_i\) is the expression value of a cell in the cell type c (e.g. UMI count or CPM), \(N\) is the number of cell types in a data set and \(C = \{cell\ type\ 1, cell\ type\ 2, ..., cell\ type\ N\}\). Note that log transformation was omitted when available data was already log transformed.We performed a one-sided test (\(\beta_E>0\)) which is essentially testing the positive relationship between cell specificity and genetic association of genes.
In principle, this model is same as tissue specificity analyses with MAGMA on SNP2GENE process where tissue specific expression was used instead of cell specific expression.
The file format of scRNA-seq data set is, Ensembl gene ID in the first column with column name "GENE", N columns for per cell type average expression and average expression across cell types with column name "Average". MAGMA gene-property analysis is run with the following command.
From FUMA v1.3.4 (MAGMA v1.07)
magma --gene-results [input file name].genes.raw \
--gene-covar [file name of selected scRNA-seq data set] \
--model condition-hide=Average direction=greater \
--out [output file name]
Until FUMA v1.3.3d (MAGMA v1.06)
magma --gene-results [input file name].genes.raw \
--gene-covar [file name of selected scRNA-seq data set] condition=Average onesided=greater \
--out [output file name]
The extension of output files from gene-property analysis using MAGMA v1.06 and v1.07 are different. Please refer to Outputs for details.
3-step workflow with multiple scRNA-seq data sets
Since integration of scRNA-seq across datasets is highly challenging due to complex batch effects, the 3-step workflow is aimed to bypass this problem by systematically compare cell type associations across datasets using conditional analyses.Step 1. per dataset cell type analysis
In the first step, MAGMA cell specificity analyses are performed for
each of the user selected datasets separately using the regression model
described in the previous section.
Multiple testing correction is applied to the results for all tested
cell type across datasets and significant cell types are retained for the
next step.
For example, when dataset A with 5 cell types and B with 10 cell types
are selected, then multiple test correction is performed for 15 tested cell types.
Note that outputs (both plots in result page and output files) also include
adjusted P-value per dataset.
Step 2. within dataset conditional analysis
The second step is a within dataset conditional analysis.
It is often the case that there are multiple similar cell types defined
in a scRNA-seq dataset, especially when the resolution of cell types is
high. The gene expression profiles of those cell types tend to strongly
correlate with each other, and when a cell type is strongly associated
with a trait it is therefore not clear whether that reflects a genuine
involvement of that cell type or whether there is confounding due to
expression in another cell type correlated with it.
In step 2, a systematical step-wise conditional analysis per dataset
is performed, by setting thresholds for proportional significance (\(PS\))
of the conditional P-value of a cell type relative to the marginal P-value as
described in the table.
\(PS\) is defined as
$$PS_{a,b}=-log10(p_{a,b})/-log10(p_a)$$
where \(p_a\) is the marginal P-value for the cell type \(a\) using the base-model
$$Z = \beta_0 + E_a\beta_{E_a} + A\beta_A + B\beta_B + \epsilon$$
and \(p_{a,b}\) is the conditional P-value of the cell type
\(a\) conditioning on the cell type \(b\) using the following model,
$$Z = \beta_0 + E_a\beta_{E_a} + E_b\beta_{E_b} + A\beta_A + B\beta_B + \epsilon$$
In summary, forward selection (retain the cell type
with the lowest marginal P-value) was performed for a pair of cell types
which were jointly explained (\(PS_{a,b}\)<0.2 and \(PS_{b,a}\)<0.2) or
one association was mainly driving the other’s (\(PS_{a,b}\)≥0.5 and \(p_{b,a}\)<0.05,
or \(PS_{a,b}\)≥0.8 and \(PS_{b,a}\)<0.5).
In the case of partially joint associations (\(PS_{a,b}\)≥0.5 and \(PS_{b,a}\)≥0.5) or
independent (\(PS_{a,b}\)≥0.8 and \(PS_{b,a}\)≥0.8), both cell types were retained.
(scenarios are ordered by the priority)
| Scenario | Cell type a | Cell type b | Cell type a state | Cell type b state | Description |
|---|---|---|---|---|---|
| 1 | \(PS_{a,b}≥0.8\) | \(PS_{b,a}≥0.8\) | indep | indep | The association of cell type \(a\) and \(b\) are independent. |
| 2 | \(p_{a,b}≥0.05\) | \(p_{b,a}≥0.05\) | join | joint-drop | The association of cell type \(a\) and \(b\) are depending each other, and the model cannot distinguish association of two cell types. In this case, cell type \(a\) is retained and \(b\) is dropped as cell type \(a\) has more significant marginal P-value, but it does not mean association of cell type \(a\) is true and \(b\) is not. |
| 3 | \(PS_{a,b}<0.2\) | \(PS_{b,a}<0.2\) | joint | joint-drop | Similar to the scenario 2, but the association of cell type \(a\) and \(b\) are not completely explained by each other. In this case, only cell type \(a\) is retained as the significance of cell type \(b\) drop to less than 20% of the marginal association. The output (state of cell types) is exactly the same as scenario 2, however there might be still some signals specific to each cell type \(a\) and \(b\). |
| 4 | \(PS_{a,b}≥0.5\) | \(p_{b,a}≥0.05\) | main | drop | The association of cell type \(b\) is completely depending on the association of cell type \(a\). Only cell type \(a\) is retained. |
| 5 | \(PS_{a,b}≥0.8\) | \(PS_{b,a}<0.2\) | main | partial-drop | The association of cell type \(b\) is mostly depending on the association of cell type \(a\) but cell type \(a\) cannot completely explain the association of cell type \(b\). In this case, only cell type \(a\) is retained as the significance of cell type \(b\) drop to less than 20% of the marginal association, however there are some amount of signals remained (since P-value is still less than 0.05). |
| 6 | \(PS_{a,b}≥0.5\) | \(PS_{b,a}≥0.5\) | partial-joint | partial-joint | The association of cell type \(a\) and \(b\) are only partially explained by each other but majority of signals are coming from the independent associations. Both cell type \(a\) and \(b\) are retained. |
| 7 | \(PS_{a,b}≥0.2\) | \(PS_{b,a}≥0.2\) | partial-joint | partial-joint-drop | Similar to scenario 6 but larger proportion of signals are explained by each other. In this case, only cell type \(a\) is retained as cell type \(b\) remain less than 20% of marginal significance, however there might still be specific underlying signal for cell type \(b\). |
| 8 | \(PS_{a,b}≥0.2\) | \(p_{b,a}≥0.05\) | partial-joint | joint-drop | The association of cell type \(b\) is completely explained by cell type \(a\) but there are part of association of cell type \(a\) dependent on cell type \(b\). In this case, only cell type \(a\) is retained. |
| 9 | \(PS_{a,b}≥0.2\) | \(PS_{b,a}<0.2\) | partial-joint | partial-joint-drop | The association of cell type \(b\) is mostly explained by cell type \(a\) but there are part of associations dependent on each other. In this case, only cell type \(a\) is retained. |
Note that when associations of two cell types are jointly explained,
only one cell type with the lowest marginal P-value is retained for the third step.
However, this does not mean the discarded cell type is less important
than the retained cell type, but the result suggests that the associations
of these two cell types cannot be distinguished.
Although conditional P-values are often proportional to marginal P-values,
it is possible that cell type with higher marginal P-value results in
less conditional P-value for a pair of cell types (i.e. \(p_{b,a}\)<\(p_{a,b}\)).
Therefore, when \(PS_{a,b}\)<0.2 and \(PS_{b,a}\)≥0.2,
the order of cell types was flipped for forward selection.
Although only retained cell types were used for the third step, the
results of within dataset conditional analyses for any pair of cell
types were further breakdown into 8 categories as described in the table.
This is to provide better understanding of the relationship of two
significantly associated cell types.
For example, in both scenario 4 and 5, cell type B is dropped and
cell type A is considered as the main driver of the association.
However, in scenario 4, association of cell type B
cannot be completely explained by cell type A as conditional P-value of
cell type B is still <0.05. Therefore, there might still be a unique
signal to cell type B, however, as large amount of significance is dropped,
the cell type B is not retained for the further step.
Note that step 2 is only performed for dataset where more than one cell types
reached significance after multiple testing correction across datasets.
Step 3. cross datasets conditional analysis
The last step is to unravel relationships between significantly associated
cell types across datasets.
Although the absolute gene expression values in different datasets are not
directly comparable, cross-datasets conditional analysis allows us to test
the extent to which the significant gene expression profiles found in
different data sets reflect the same or similar association signals.
The analysis is performed for all possible cross-dataset pairs of
significant cell types retained from the second step.
Then the \(PS\) of the cross-datasets (CD) conditional P-value of a cell type
relative to the CD marginal P-value is computed for each cell type of all possible pairs.
For each pair of cell types from different datasets, the following
three regression models were tested to incorporate the effect of
the average expression from the other dataset:
$$Z=\beta_0 + E_c1\beta_{E_{c1}} + A_1\beta_{A_1} + A_2\beta_{A_2} + B\beta_B + \epsilon$$
$$Z=\beta_0 + E_c2\beta_{E_{c2}} + A_1\beta_{A_1} + A_2\beta_{A_2} + B\beta_B + \epsilon$$
$$Z=\beta_0 + E_c1\beta_{E_{c1}} + E_c2\beta_{E_{c2}} + A_1\beta_{A_1} + A_2\beta_{A_2} + B\beta_B + \epsilon$$
where \(E_{cx}\) is an average log transformed expression of cell type \(c\) from
dataset \(x\), and \(A_x\) is an average expression across cell types in dataset \(x\).
In this step, we define P-value of testing alternative hypothesis \(\beta_{E_{cx}}>\)0
from 1st and 2nd models as CD marginal P-value,
and \(\beta_{E_{cx}}\)>0 from 3rd model as CD conditional P-value for a cell type \(c\)
from a dataset \(x\).
Note that, when associations of two cell types from
different datasets with a trait are largely disappeared by conditioning
on each other, it suggests that associations of those cell types were
driven by similar genetic signals but this does not measure the similarity
of two cell types (i.e. it cannot be concluded that the cell types from
the different datasets are the same).
Note that step 3 is only performed where there are significant cell types from
more than one datasets.
Please be aware that, some of scRNA-seq, there are multiple datasets
available from a single scRNA-seq data resource.
For example, Tabula Muris FACS data, one dataset contains all cell types from all 20 tissues,
and there are 20 datasets for each tissue separately.
When both TabulaMuris_FACS_all and TabulaMuris_FACS_Aorta datasets are selected, for instance,
exact same cell types in the Aorta dataset exist in the dataset with all tissues.
Testing both datasets are still relevant as average expression across cell types is different for each dataset,
however, step 3 is not relevant in this case as they are exactly the same cell type.
When step 3 is activated, FUMA will still perform all possible pair of
significant cell types across datasets.
However the pair of exact same cell type will be collinear (MAGMA outputs NA for such pairs).
Data sets
Starting from FUMA version 1.7.0 with the update of new datasets to FUMA Cell type, note the following changes:- Scripts for pre-processing the batch of scRNAseq datasets in Phung et al. 202x is hosted at https://github.com/tanyaphung/scrnaseq_viewer.
-
The pre-processed scRNAseq data within FUMA can be downloaded in the Download page (scroll to the section FUMA Cell Type).
Pre-process was performed as the following steps. Please see each script for more details.
- When the obtained value was the read count, the count was converted into the count per million (CPM) to allow correction for the total number of reads per cell.
- QC of cells was performed as described in the original study unless the obtained data was already QCed.
- Cells with uninformative cell type labels (e.g. ‘unclassified’ or ‘unknown’) were excluded, unless specified.
- The expression value (UMI count, CPM, RPKM or TPM) was log2 transformed with pseudo-count 1 (unless the it's already done) and per gene per cell type average was computed. When there were multiple levels of cell type labels, the average expression was computed for each level separately.
- Genes provided in the processed datasets were mapped to human Ensembl gene ID (v92 GRCh37).
| Data name | Link | Description | Reference | Last update |
|---|---|---|---|---|
| Adult Human Brain | Data: https://cellxgene.cziscience.com/collections/283d65eb-dd53-496d-adb7-7570c7caa443 | Adult Human Brain. Brief summary: In Siletti et al. 2023, postmortem tissues were isolated from 3 donors. Neurons were enriched from ~100 locations across the forebrain (cerebral cortex, hippocampus, cerebral nuclei, hypothalamus, and thalamus), midbrain, and hindbrain (pons, medulla, and cerebellum). In total, there were ~3M cells that formed 31 superclusters, 461 clusters, and 3313 subclusters. From the data downloaded from cellxgene, we processed 105 datasets that we categorized to different specific regions of the brain. For each of the 105 datasets, we created two files for 2 level of cell type annotations. For more information on how this data was processed, please check: https://github.com/tanyaphung/scrnaseq_viewer/blob/main/notes/14_Silletti_adult_human_brain_notes.md |
Siletti et al. 2023. Transcriptomic diversity of cell types across the adult human brain.
Science. Vol 382, Issue 6667. PMID: 36318921 |
26 January 2025 |
| GSE168408 | Website: http://brain.listerlab.org/, Data: https://console.cloud.google.com/storage/browser/neuro-dev/Processed_data;tab=objects?prefix=&forceOnObjectsSortingFiltering=false | Human Prefrontal Cortex. 26 postmortem prefrontal cortex samples spanning 6 stages: Fetal, Neonatal, Infancy, Childhood, Adolescence, and Adult resulting in 154,738 single nuclei. 3 levels of annotation: level 1 consists of 3 cell types, level 2 consists of 18 cell types, and level 3 consists of 86 cell types. From 26,747 genes, 26,671 genes were mapped to hs ENSG ID. In total, 18 data sets were created (6 stages for each of the 3 levels) |
Herring et al. 2022. Human prefrontal cortex gene regulatory dynamics from gestation to adulthood at single-cell resolution.
Cell. 185, 4428-4447. PMID: 36318921 |
19 December 2022 |
| Tabula Muris | FACS: https://figshare.com/articles/Single-cell_RNA-seq_data_from_Smart-seq2_sequencing_of_FACS_sorted_cells_v2_/5829687 , droplet: https://figshare.com/articles/Single-cell_RNA-seq_data_from_microfluidic_emulsion/5715025 | Multiple tissues/organs of mouse samples. FACS: From 53,760 cells in the raw read count matrix, 44,949 cells exist in the annotation file. Cells with label "unknown" were included as it was stated in the original study that they are potential novel cell types. From 23,433 genes, 15,131 genes were mapped to hs ENSG ID. In total, 22 data sets were created (1 for cell types from all tissues/organs together, 20 for each tissue/organ separately and 1 for all Brain cell types (including both Brain_Meyloid and Brain_Non-Meyloid)). droplet: From 2,990,808 cells in the raw read count matrix, 54,837 cells exist in the annotation file. Cells with label "unknown" were included as it was stated in the original study that they are potential novel cell types. From 23,433 genes, 15,131 genes were mapped to hs ENSG ID. In total, 13 data sets were created (1 for cell types from all tissues/organs together and 12 for each tissue/organ separately). |
The Tabula Muris Consortium et al. 2018.
Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris.
Nature. 562, 367-372. PMID: 30283141 |
4 Feb 2019 |
| Mouse Cell Atlas (GSE108097) | Website: http://bis.zju.edu.cn/MCA/, Data: https://figshare.com/s/865e694ad06d5857db4b | Multiple tissues/organs of mouse samples. A file "Figure2-batch-removed.txt.gz" was used in which batch was removed and cells were already QCed. 61,637 cells were available and not additional filtering was performed. From 25,133 genes, 15,640 genes were mapped to hs ENSG ID. In total 37 data sets were created as the following; 1) all tissues/developmental stages together (731 unique cell types), 2) only adult mouse samples (437 cell types from 18 tissue), 3) only embryo samples (including fetal tissues, 137 cell types), 4) only neonatal samples (108 cell types), 5-37) per tissue per sample type (adult, embryo, neonatal and cell line, 33 combination in total). |
Han et al. 2018.
Mapping the Mouse Cell Atlas by Microwell-Seq. Cell. 172, 1091-1107. PMID: 29474909 |
17 July 2018 |
| Allen Brain Atlas Cell Type |
Human LGN: http://celltypes.brain-map.org/api/v2/well_known_file_download/694416667 , MTG: http://celltypes.brain-map.org/api/v2/well_known_file_download/694416044 Mouse new version ALM2: http://celltypes.brain-map.org/api/v2/well_known_file_download/694413179 , LGd2: http://download.alleninstitute.org/informatics-archive/current-release/rna_seq/mouse_LGd_gene_expression_matrices_2018-06-14.zip , VISp2: http://celltypes.brain-map.org/api/v2/well_known_file_download/694413985 Mouse old version ALM: https://portals.broadinstitute.org/single_cell/study/a-transcriptomic-taxonomy-of-adult-mouse-visual-cortex-visp , LGp: https://portals.broadinstitute.org/single_cell#study-a-transcriptomic-taxonomy-of-adult-mouse-anterior-lateral-motor-cortex-alm , VISp: https://portals.broadinstitute.org/single_cell#study-a-transcriptomic-taxonomy-of-adult-mouse-lateral-geniculate-complex-lgd |
Human and mouse brain samples. For each data, level 1, 2 and 3 cell types were processed separately. For Human MTG, LGN and mouse ALM2, LGd2 and VISp2, sum of read counts for exon and introns were computed for each gene, to obtain gene level read counts. Human MTG: From 15,928 cells, 325 with "no class" were excluded, resulted in 15,603 cells. From 50,281 genes, 29,115 genes were mapped to unique ENSG ID. Human LGN: From 1,576 cells, 23 with "no class" were excluded, resulted in 1,553 cells. From 50,281 genes, 29,115 genes were mapped to unique ENSG ID. Mouse ALM2: From 10,068 cells, 99 which "Low Quality" and 396 cells with "no class" were excluded, resulted in 9,573 cells. From 45,768 genes, 16,093 genes were mapped to unique hs ENSG ID. Mouse LGd2: From 1,996 cells, 122 with "Outlier" were excluded, resulted in 1,874 cells. From 45,768 genes, 16,093 genes were mapped to unique hs ENSG ID. Mouse VISp2: From 15,413 cells, 490 with "Low Quality" and 674 cells with "no class" were excluded, resulted in 14,249 cells. From 45,768 genes, 16,093 genes were mapped to unique hs ENSG ID. Mouse ALM: All 1,301 cells were used. From 45,764 genes, 16,068 genes were mapped to unique hs ENSG ID. Mouse LGd: From 1,827 cells, 17 cels with label "Outlier" were excluded, resulted in 1,810 cells. From 45,761 genes, 15,837 genes were mapped to unique hs ENSG ID. Mouse VISp: All 1,679 cells were used. From 24,057 genes, 15,097 genes were mapped to unique hs ENSG ID. |
(For Human MTG) Hodge, et al. 2018. Conserved cell types with divergent features between human and mouse cortex. bioRxiv. doi: https://doi.org/10.1101/384826 (For Mouse VISp2 and ALM2) Tasic et al. 2018. Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72-78. PMID: 30382198 (For Mouse VISp data set) Tasic et al. 2016. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci. 19, 335-346. PMID: 26727548 |
4 Feb 2019 |
| DropViz | http://dropviz.org/ | Mouse brain samples. "Metacells" data downloaded from DropViz website was used which is the aggregated data per 565 sub-cluster not the individual cell level UMI counts. The UMI was the sum of all the cells in a subcluster, therefore we converted to CPM. In the annotation of each sub-cluster, "class" column was used as level 1 cell type and subcluster was used as level 2. From 32,307 genes, 16,097 genes were mapped to hs ENSG ID. |
Arpair et al. 2018. Molecular diversity and specializations among the cells of adult mouse brain. Cell. 9, 1015-1030. PMID: 30096299 | 4 Feb 2019 |
| DroNc | Human: https://www.gtexportal.org/ / https://portals.broadinstitute.org/single_cell#study-dronc-seq-single-nucleus-rna-seq-on-human-archived-brain, Mouse: https://portals.broadinstitute.org/single_cell#study-dronc-seq-single-nucleus-rna-seq-on-mouse-archived-brain | Human and mouse brain samples. Human: Expression data was downloaded from GTEx website (also available from Broadinstitute Single Cell Portal). Cells with cluster 1-14 or 16 were used since those clusters were assigned in the original study. The cell type label was manually assigned to the cluster index based on the figure 2a in the original paper. From 14,963 cells, 14,137 cells were used. From 32,111 genes, 31,852 genes were mapped to ENSG ID. Mouse: Cells with label "Unclassified", "Doublets" or "ChP" were excluded as they are not assigned in the original study. From 13,313 cells, 11,148 cells were used. From 17,3080 genes, 13,335 genes were mapped to hs ENSG ID. |
Habib et al. 2017. Massively parallel single-nucleus RNA-seq with DroNc-seq.
Nat. Methods. 14, 955-958. PMID: 28846088 |
17 July 2018 |
| Mouse Brain Atlas (Linnarsson's lab) | http://mousebrain.org/ | Mouse brain samples. Five expression matrices were obtained for level 5, level 6 rank 1-4. Note that the expression value was already aggregated per cell type and we did not use individual cell level expression data. Each of 5 data sets were processed separately. From 27,997 genes, 16,420 genes were mapped to hs ENSG ID. |
Zeisel et al. 2018. Molecular architecture of the mouse nervous system. Cell. 9 999-1014. PMID: 30096314 | 4 Feb 2019 |
| GSE59739 (Linnarsson's lab) | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE59739 | Mouse brain samples (dorsal root ganglion L4-L6 from 6-8 weeks old mice). Expression data was obtained from GEO and annotation of each cell was extracted from family soft file. Cells with label NF, NP, PEP or TH in Level 1 cell types were used to be consistent with the original study. From 865 cells in the expression data, 622 cells were used. From 25,333 genes, 15,084 genes were mapped to hs ENSG ID. Per cell type average expression was computed for level 1, 2 and 3 separately. |
Usoskin et al. 2015. Unbiased classification of sensory neuron types by large-scale single-cell RNS sequencing.
Nat. Neurosci. 18, 145-153. PMID: 25420068 |
17 July 2018 |
| GSE60361 (Linnarsson's lab) | https://storage.googleapis.com/linnarsson-lab-www-blobs/blobs/cortex | Mouse brain samples (cortex and hippocampus from P22-P32 mice). 3,005 cells were available. From 19,972 genes, 15,161 genes were mapped to hs ENSG ID. Per cell type average expression was computed for level 1 and level 2 separately. For level 2, 189 cells with label "none" were excluded. |
Zeisel et al. 2015. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq.
Science. 347, 1138-1142. PMID: 25700174 |
17 July 2018 |
| GSE75330 (Linnarsson's lab) | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE75330 | Mouse brain samples (oligodendrocytes from day21-90 mice). 5,069 cells were available. From 23,556 genes, 15,816 genes were mapped to hs ENSG. |
Marques et al. 2016.
Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system.
Science. 352, 1326-1329. PMID: 27784195 |
17 July 2018 |
| GSE78845 (Linnarsson's lab) | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE78845 | Mouse brain samples (stellate and thoracic sympathetic ganglia from postnatal day 27-33 mice). Cells with label "unclassified" were excluded. From 298 cells, 213 cells were used. From 16,892 genes, 13,804 genes were mapped to hs ENSG ID. |
Furlan et al. 2016. Visceral motor neuron diversity delineates a cellular basis for nipple-and plio-erection muscle control.
Nat. Neurosci. 19, 1331-1340. https://www.ncbi.nlm.nih.gov/pubmed/27571008 |
17 July 2018 |
| GSE76381 (Linnarsson's lab) | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE76381 | Human brain samples (ventral midbrain from 6-11 weeks embryos) and mouse brain samples (ventral midbrain from E11.5-E18.5 embryos). Only human embryo (1,977 cells) and mouse embryo (1,907 cells) data set were used. Cells with label "Unk" (unknown) were excluded. For human, from 1,977 cells, 1695 cell were used. From 19,531 genes, 16,885 genes were mapped to ENSG ID. For mouse, from 1,907 cells, 1,518 cells were used. From 24,378 genes, 15,826 genes were mapped to hs ENSG ID. |
La Manno et al. 2016. Molecular diversity of midbrain development in mouse, human, and stem cells.
Cell. 167, 556-580. PMID: 27716510 |
17 July 2018 |
| GSE95752, GSE95315 and GSE104323 (Linnarsson's lab) | GSE95752: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE95752, GSE95315: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE95315, GSE104323: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE104323 | Mouse brain samples (dentate gyrus from P5-P26 and P50-P65 for GSE95752, P12-P35 for GSE95315 and E16.5 and P0-P132 for GSE104323). GSE95752: 2,303 cells were available. From 16,131 genes, 143,470 genes were mapped to hs ENSG ID. GSE95315: Cell types obtained from family soft file (41 types) are merged into 22 cluster as presented in the original study. 5,454 cells were available. From 14,545 genes, 12,640 genes were mapped to hs ENSG ID. GSE104323: From 24,216 cells, 24,185 cells with valid cell labels were used (cells with blank in the cell type column were excluded). From 27,933 genes, 16,146 genes were mapped to hs ENSG ID. |
Hochgerner et al. 2018. Conserved properties of dentate gyrus neurogenesis across postnatal development revealed by single-cell RNA sequencing.
Nat. Neurosci. 21, 290-299. PMID: 29335606 |
17 July 2018 |
| GSE101601 (Linnarsson's lab) | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE101601 | Human brain samples (Temporal cortex from post-mortem samples) and mouse brain samples (Somatosensory cortex from postnatal days 21-37 mice). Human (2,028 cells) and mouse (2,192 cells) data sets were processed separately. For human, from 28,274 genes, 21,459 genes were mapped to ENSG ID. For mouse, from 24,339 genes, 15,826 genes were mapped to hs ENSG ID. |
Hochgerner et al. 2017. STRT-seq-2i: dual-index 5' single cell and nucleus RNA-seq on an addressable microwell array.
Sci. Rep. 7: 16327. PMID: 29180631 |
17 July 2018 |
| GSE74672 (Linnarsson's lab) | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE74672 | Mouse brain samples (hypothalamus from postnatal days 14-28 mice). Only 2881 cells were available in the expression file, though it was mentioned that 3131 cells in the original paper. From 24,341 genes, 15,826 genes were mapped to hs ENSG ID. Per cell type average expression was computed for level 1 and 2 separately. Level 2 label was only available for neurons. From 898 neurons, 126 cells with level 2 label "uc" (unclassified) were excluded. |
Romanov et al. 2017. Molecular interrogation of hypothalamic organization reveals distinct dopamine neuronal subtypes.
Nat. Neurosci. 20, 176-188. PMID: 27991900 |
17 July 2018 |
| GSE67602 (Linnarsson's lab) | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE67602 | Mouse epidermis from dorsal skin (~8 weeks). 1,422 cells were available. From 25,932 genes, 15,802 genes were mapped to hs ENSG. |
Joost et al. 2016. Single-cell transcriptomics reveals that differentiation and spatial signatures shape epidermal and hair follicle heterogeneity.
Cell Syst. 3, 221-237. PMID: 27641957 |
17 July 2018 |
| GSE103840 (Linnarsson's lab) | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE103840 | Mouse brain samples (dorsal horn from 3-4 weeks old mice). 1,545 cells were available. From 24,378 genes, 15,826 genes were mapped to hs ENSG ID. |
Haring et al. 2018. Neuronal atlas of the dorsal horn defines its architecture and links sensory input to transcriptional cell types.
Nat. Neurosci. 21, 869-880. PMID: 29686262 |
17 July 2018 |
| GSE87544 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE87544 | Mouse brain samples (hypothalamus from 8-10 weeks l=old mice). From 14,437 cells, 6,507 cells with condition "Normal" were extracted. Cells with label "zothers" were further excluded resulted in 5,350 cells. To be consistent with the original study, cells with <=2000 genes expressed (0 expression) were excluded. In total, 1,039 cells were used. From 23,284 genes, 15,116 genes were mapped to hs ENSG ID. In the original study, there are 45 cell types but in the downloadable data there was no NFO but instead IMO and SCO. By checking with the authors, IMO (immature oligodendrocyte) = NFO and SCO (Subcommissural organ) is extra. |
Chen et al. 2017. Single-cell RNA-seq reveals hypothalamic cell diversity.
Cell Rep. 18, 3227-3241. PMID: 28355573 |
17 July 2018 |
| GSE98816 and GSE92235 | GSE98816: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE98816, GSE92235: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE92235 | Mouse brain vascular cells (GSE98816) and lung vascular cells (GSE92235) from 10-19 weeks old mice. Cell type label was obtained directly from the authors by requesting. GSE98816: 3,186 cells were available. From 19,937 genes, 15,302 genes were mapped to hs ENSG ID. GSE92235: 1,504 cells were available. From 21,948 genes, 15,801 genes were mapped to hs ENSG ID. |
Vanlandewijck et al. 2018. A molecular atlas of cell types and zonation in the brain vasculature.
Nature. 554, 475-480. PMID: 29443965 |
17 July 2018 |
| GSE81547 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE81547 | Human pancreas samples (healthy donors between 1 month to 54 years old). 2,544 cells were available. Cells with label "unsure" was defined as "PP" in the original study. From 23,465 genes, 20,706 genes were mapped to hs ENSG ID. |
Enge et al. 2017. Single-cell analysis of human pancreas reveals transcriptional signatures of ageing and somatic mutation patterns.
Cell. 171, 321-330. PMID: 28965763 |
17 July 2018 |
| GSE104276 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE104276 | Human brain samples (prefrontal cortex from 8-26 weeks after gestation). 2,309 cells were available. From 24,153 genes, 21,177 genes were mapped to ENSG ID. Two data sets were created; 1) per cell type average across different ages, 2) per cell type per age average expression. |
Zhong et al. 2018. A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex.
Nature. 555, 524-528. PMID: 29539641 |
17 July 2018 |
| GSE82187 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE82187 | Mouse brain samples (striatum from 5-7 weeks old mice). Only microfluid data was used since FACS data was limited to neurons. From 1,208 cells, 705 cells from microfluid were used. From 18,840 genes, 14,189 genes were mapped to hs ENSG ID. |
Gokce et al. 2016. Cellular taxonomy of the mouse striatum as revealed by single-cell RNA-seq.
Cell Repo. 16, 1126-1137. PMID: 27425622 |
17 July 2018 |
| GSE89232 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE89232 | Human blood samples. 957 cells were available. From, 20.689 genes, 17,035 genes were mapped to hs ENSG ID. |
Breton et al. 2016. Human dendritic cells (DCs) are derived from distinct circulating precursors that are precommitted to become CD1c+ or CD141+ DCs.
J. Exp. Med. 213, 2861-2870. PMID: 27864467 |
17 July 2018 |
| GSE100597 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE100597 | Mouse embryos (E3.5, E4.5, E5.5 and E6.5). Developmental stage was used as cell label. 721 cells were available. From 24,83 genes, 14,513 genes were mapped to hs ENSG ID. |
Mohammed et al. 20174. Single-cell landscape of transcriptional heterogeneity and cell fate decisions during mouse early gastrulation.
Cell Repo. 20, 1215-1228. PMID: 28768204 |
17 July 2018 |
| GSE93374 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE93374 | Mouse brain samples (hypothalamic arcuate-median eminence complex from 4-12 weeks old mice). Cells with label "miss" in the column "clust_all" were excluded. From 21,086 cells, 20,921 cells were used. Level 1, level 2 and clusters for neurons were processed separately resulted in three data sets. For clusters for neurons, non-neuronal cells were excluded (with label "miss" in "clust_neurons" column; 13,079 neuronal cells in total). From 19,743 genes, 14,366 genes were mapped to hs ENSG ID. |
Campbell et al. 2017. A molecular census of arcuate hypothalamus and median eminence cell types.
Nat. Neurosci. 20, 484-496. PMID: 28166221 |
17 July 2018 |
| GSE92332 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE92332 | Mouse small intestine epithelium samples (7-10 weeks old mice). Expression data was obtained for SMATRseq (1,522 cells) and droplet (7,216 cells) data set. Each data set was processed separately. For SMARTseq, from 20,108 genes, 14,714 genes were mapped to hs ENSG ID. For droplet, from 15,971 genes, 12,865 genes were mapped to hs ENSG ID. |
Haber et al. 2017. A single-cell survey of the small intestinal epithelium.
Nature. 551, 333-339. PMID: 29144463 |
17 July 2018 |
| GSE89164 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE89164 | Mouse brain samples (hindbrain from P0 mice). Two count matrices for mouse replicates were combined and extracted 4366 cells exist in the cluster information. Cell label was manually assigned to the cluster index based on the original study. From 20,648 genes, 13,176 genes were mapped to hs ENSG ID. |
Alies et al. 2017. Cell fixation and preservation for droplet-based single-cell transcriptomics.
BMC Biol. 15: 44. PMID: 28526029 |
17 July 2018 |
| GSE67835 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE67835 | Human brain samples (cortex from adult and fetal samples). Two data sets with and without fetal sample were created. 466 cells were available (of which 135 cells were fetal samples). From 22,088 genes, 19,749 genes were mapped to ENSG ID. |
Darmanis et al. 2015. A survey of human brain transcriptome diversity at the single cell level.
Proc. Natl. Acad. Sci. USA. 112, 7285-90. PMID: 26060301 |
17 July 2018 |
| GSE106678 | https://portals.broadinstitute.org/single_cell/study/snucdrop-seq-dissecting-cell-type-composition-and-activity-dependent-transcriptional-state-in-mammalian-brains-by-massively-parallel-single-nucleus-rna-seq | Mouse brain samples (cortex from 6-10 weeks old mice). Expression data was obtained from Broadinstitute Single Cell Portal. 18,194 cells were available. From 30,341 genes, 15,782 genes were mapped to hs ENSG ID. |
Hu et al. 2017. Dissecting cell-type composition and activity-dependent transcriptional state in mammalian brains by massively parallel single-nucleus RNA-seq.
Mol. Cell. 68, 1006-1015. PMID: 29220646 |
17 July 2018 |
| GSE84133 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE84133 |
Human and mouse pancreas samples. Humam: Human sample 4 was excluded as it is with sample status T2D. All 7,266 cell were used and from 20,125 genes 19,546 genes were mapped to unique ENSG ID. Mouse: All 1,886 cells were and from 14,878 genes, 12,741 genes were mapped to unique hs ENSG ID. |
Baron, et al. 2016. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure.
Cell Systems 3, 346-360. PMID: 27667365 |
4 Feb 2019 |
| 10x PBMC | https://community.10xgenomics.com/t5/Data-Sharing/10x-Single-Cell-3-Paper-Zheng-et-al-2016-Datasets/td-p/231 |
Human peripheral blood mononuclear cells (PBMCs). Cell label was downloaded from github repository https://github.com/10XGenomics/single-cell-3prime-paper. Data set was downloaded from 10X website directory. All 68,579 cells were used. Genes were annotated to ENSG ID in the original data (32,738 genes). |
Zheng. et al. 2017. Massively parallel digital transcriptional profiling of single cells.
Nat. Communs. 8, 14049. PMID: 28091601 |
4 Feb 2019 |
| PsychENCODE | http://resource.psychencode.org/ |
Human developmental and adult brain samples. For developmental dataset, 4,249 cells were available. For adult dataset, from 27,412 cells, 32 cells with cell type label NA were excluded, resulted in 27,380 cells. From 15,086 and 17,176 genes, 15,019 and 16,243 genes were mapped to unique ENSG ID for developmental and adult datasets, respectively. |
Wang. et al. 2018. Comprehensive functional genomic resource and integrative model for the human brain.
Science. 362, eaat8464. PMID: 30545857 |
19 May 2019 |
| GSE97478, GSE106707 |
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE97478
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE106707 |
Mouse striatum and cortex samples. GSE97478: 1,122 cells were available. From 12,936 genes, 11,299 genes were mapped to unique hs ENSG ID. GSE106707: 3,417 cells were available. From 10,002 genes, 8,940 genes were mapped to unique hs ENSG ID. |
Muoz-Manchado. et al. 2018. Diversity of interneurons in the dorsal striatum revealed by single-cell RNA sequencing and PatchSeq.
Cell Rep. 24, 2179-2190.e7. PMID: 30134177 |
19 May 2019 |
Publish results
How to publish FUMA results
1. Prepare jobs in SNP2GENE and GENE2FUNC
You can publish any of existing SNP2GENE job in your account but only the ones without any error. MAGMA results are optional but it's highly recommended to include them too. You can also publish GENE2FUNC results together with SNP2GENE results if the GENE2FUNC job is performed for mapped genes from the corresponding SNP2GENE job.2. Publish results
You can publish your results from your job list on SNP2GENE page. There is a "publish" button for each SNP2GENE job.
When you click the "publish" button, a popup will open where you can specify some features of the job. Please fill the features in the table below as much as possible before submit your job.
| Features | Description |
|---|---|
| Selected SNP2GENE jobID | Auto filled when you click the "publish" button. This value is not changeable. |
| Corresponding GENE2FUNC jobID | Auto filled when there is a recognized GENE2FUNC job. FUMA recognizes a matched GENE2FUNC job only when the GENE2FUNC job has been performed by using "GENE2FUNC" button (internal submission). If you manually submit GENE2FUNC for the corresponding SNP2GENE job, you can manually specify here. |
| Title | Title of the published job should be self-descriptive, although it is auto filled by the title of the selected SNP2GENE job. If the title is not clear enough, the developer might contact you to provide a sufficient information. |
| Author | Auto filled with the user name but please provide your full name. |
| Auto filled with your registered email address. Please provide an email address that is reachable to you. Any future modification/deletion of the published job will be only processed when it is requested by the matched email. | |
| Phenotype | Please provide phenotype of the GWAS if applicable. |
| Publication | This is the publication where the selected SNP2GENE job is described (not the reference to the summary statistics). This can be any format as long as users are able to find the publication. Please provide PubMed ID if possible (e.g. PMID: 29184056). If you don't have publication yet, please let the developer know once the publication becomes available. You can also provide preprint DOI. |
| Link to summary statistics | If the summary statistics used in this job is publicly available, please provide the original link. |
| Reference of summary statistics | This should be the original publication of the summary statistics. This can be same as the publication above when a new GWAS result is presented in the publication. |
| Notes | You can provide any additional information here (max 300 characters). For example, when there are multiple summary statistics available from the same study, you should specify which result this is referring to. |
3. Check your published results
Published job will be listed in the "Browse Public Results" page. Please have a look at your published job to check if there is any problem.
Modify/delete published result
Modification/deletion is only possible when the user is logged in with the same email address as the entry.
Users' responsibility
What other users can do?
The "Browse Public Result" page does not require users to login.